|
| CHIANTI |
| An Atomic Database for Spectroscopic Diagnostics of Astrophysical Plasmas |
| USER GUIDE - Version 4.0 for the Version 4 database - 20 Sep 2002 |
This document describes the general characteristics of the new Version 4.0 of the CHIANTI package (database and IDL procedures).
A considerable number of new features, both in the database and the software, have been added (see Young et al., 2002, ApJ submitted) . We have tried to keep a distinction between the previous and the new features in this document.
Users of previous versions, please NOTICE:
The main new features described in this guide are
Backward compatibility has been assured for almost all cases. In practice, previous users of CHIANTI will be able to use the new V.4 software as before, with just minor changes. On the other hand, software versions prior to 4 are not compatible with the v.4 database.
This document replaces the previous CHIANTI guide (Version 1.0).
Written by Giulio Del Zanna (DAMTP), 15 August 2002, with contributions from Peter Young (RAL) and the other CHIANTI team members.
CHIANTI is a collaborative project involving the Naval Research Laboratory (Washington DC, USA), the Universities of Florence (Italy) and Cambridge (UK) and the Rutherford Appleton Laboratory (RAL, UK).
The CHIANTI package consists of a critically evaluated set of atomic data (energy levels, wavelengths, radiative transition probabilities and excitation data) for a large number of ions of astrophysical interest. It also includes a number of ancillary data and a suite of Interactive Data Language (IDL) programs to calculate optically thin synthetic spectra and to perform spectral analysis and plasma diagnostics.
Plasma emission codes have long been used to study UV and X-ray spectral lines emitted from solar or stellar atmospheres. A comparison of the theoretical line intensities with the observed intensities allows a determination of the physical parameters for the plasma (cf Mason and Monsignori Fossi, 1994 and Del Zanna, Landini and Mason, 2002).
It is very important, now that high accuracy atomic data are available, to improve and keep up-to-date the plasma codes.
The CHIANTI database has been used extensively by the astrophysical and solar communities to analyse emission line spectra from astrophysical sources.
The CHIANTI package is freely available at one of the CHIANTI homepages:
or at
The continued development of the CHIANTI database is dependent on continued funding which is generally available if we can demonstrate that the CHIANTI database is of use to astrophysical research. If you use CHIANTI, we only ask that you acknowledge it appropriately in any publications:
Write in the text of any publication the reference to the CHIANTI paper associated with the particular VERSION you have used:
We would appreciate if you also write in the acknowledgements of any
publication the following:
CHIANTI is a collaborative project involving NRL (USA), RAL (UK), and the Universities of Florence (Italy) and Cambridge (UK).
If a detail work on particular data is done, it would be appropriate to also refer to the paper where the original atomic calculations are detailed. References can be found at the end of each data file or on the WWW.
CHIANTI data are included into other databases. It would be appropriate to make that clear to the users so they can trace back the results they use to the original calculations.
Users should be aware of what is included in the database, of the approximations applied, and of the atomic data used. The CHIANTI results should not be blindly considered valid in all cases. For example, the CHIANTI predicted emissivities should not be used when considering temperatures outside of the validity ranges.
Any contributions and suggestions to CHIANTI are welcomed. We would appreciate a short description of how you employ CHIANTI.
The CHIANTI project was originally set up by Dr. Ken Dere of the Naval Research Laboratory (Washington, USA), Dr. Helen Mason of the Department of Applied Mathematics and Theoretical Physics at the University of Cambridge (UK), and Dr. Brunella Monsignori-Fossi of the Arcetri Astrophysical Observatory (Florence, Italy). Former students of Dr. Monsignori-Fossi (Dr. Enrico Landi) and Dr. Mason (Dr. Peter Young) helped in the creation of the database. The sad and unexpected death of Dr. Monsignori-Fossi in January 1995, led to Prof. Massimo Landini, a close associate of Dr. Monsignori-Fossi, becoming a new CHIANTI representative (University of Florence).
Additional collaborations have involved Dr. Dave Pike of the Rutherford Appleton Laboratory (RAL), who has written CHIANTI routines to run within the environment of the SOHO/CDS software (and within SolarSoft), and with Dr. Gordon Bromage, Dr. Barbara Bromage and her former student Dr. Giulio Del Zanna of the University of Central Lancashire.
Dr. Enrico Landi, now at the Naval Research Laboratory, Dr. Peter Young, now at RAL, and Dr. Giulio Del Zanna, now at DAMTP, have continued to be active collaborators in the CHIANTI project.
Young et al. (1998) used the CHIANTI database for a detailed comparison with observed EUV solar spectra to assess the diagnostic accuracy of the two data sets.
In November 2000 we have released a whole new CHIANTI package under SolarSoft.
The fitting procedure for excitation data, both electrons and protons, has been extended to allow 9 point spline fits in addition to the previous 5 point spline fits. This allows higher quality fits to data from close-coupling calculations where resonances can lead to significant structure in the thermally-averaged collision strengths.
With the addition of H I, He I and N I, the first neutral species have been added to CHIANTI.
Many existing ion data-sets have been updated, in particular most ions of the nitrogen and beryllium isoelectronic sequences. Also, new ions have been added, including Ar IV, Fe VI and Ni XXI.
The continuum routines have been re-written, including a new relativistic free-free continuum, a new free-bound, and a new two-photon continuum. New software has been written.
Minor releases of the database and the software normally include fixes and might occur a few times per year.
chianti_help@halcyon.nrl.navy.mil
The atomic data will continue to be updated regularly as new data are calculated or measured in the laboratory.
It is intended that these atomic data can be accessed and transfered into users own analysis programs, for more sophisticated applications.
The database has a tree structure, with the top directory designated with the IDL system variable !xuvtop (and named dbase within SolarSoft):
dbase/
In the top directory are the following files:
README_CHIANTI with the description of the current version.
VERSION with the version number.
Then, there is a series of subdirectories, one for each element present in the database.
Each element has a subdirectory for each ion.
The filename prefix for each ion follows spectroscopic notation.
For example, for He, we have He I and He II subdirectories:
he/
he_1/
he_2/
Then, we have a series of ancillary data that are contained in
various subdirectories:
masterlist/
has the list of the ions currently present
in the database
abundance/ with elemental abundance files.
continuum/ contains files for the continuum calculations.
dem/ has DEM files.
ioneq/ contains ionization fraction files.
ip/ has ionization potentials.
ancillary_data/instrument_responses/ with effective areas.
There are five primary ASCII files for each ion subdirectory. For example, for Fe XIV we have:
Specifies the energy levels in cm-1 and Rydbergs. It includes both experimental data and theoretical values of the levels energies.
The energy levels are obtained from NIST. Where necessary, these are supplemented by other laboratory and theoretical values.
Contains the wavelengths, gf and A values of the transitions and the indices initial and final level corresponding to the indices of the levels as given in the fe_14.elvlc file. Wavelengths calculated from the theoretical energies are of an indeterminate accuracy and their values are presented as negative values of the calculated wavelength. The `observed' wavelengths in these files are based on the experimental energies and should be the best available.
The radiative data are taken from published literature and where necessary, supplemented by new calculations.
contains the point spline fits to electron collision strengths scaled according to the the rules formulated by Burgess and Tully (1992), as described in Dere et al. (1997). All the atomic data in the CHIANTI database have been visually displayed and assessed for accuracy and any sporadic errors which sometimes creep into published results.
Accurate replication of the temperature averaged collision strength over a wide range of temperatures can be accomplished with the data in this file.
This is analogous to the fe_14.splups file, and contains the point spline fits to proton collision strengths.
The basic structure of the files is to put the data at the beginning of the file followed by comments. The original sources are documented in each data file, where also additional and detailed comments written by the CHIANTI member that assessed that particular ion can be found. You can have direct access to the references via the WWW pages.
Some additional data files are needed in various calculations. The software allows the selection of these files, from either a 'standard' selection provided within the database, or by using user defined files that are included in the current working directory, provided they have the proper file extension. For example, it is possible to create a user defined 'myfile.dem'. If the file is in the working directory, then the file will automatically be appended to the list of available DEMs from the CHIANTI database. In other cases, it is possible to select the file by using a widget that allows the user to change directory.
Any user-defined file must have the same format as those already provided (also including a 'comment' section at the end of the file)
The list of the ions present in the database
A !xuvtop/masterlist/masterlist.ions file keeps the current list of all the ions in the database. This list is used as default by many routines (for example those that calculate line intensities).
In some cases, it is possible to instead use a user-defined list of ions, to speed the calculation, or to directly supply the routines with a list of ions, via the SNGL_ION keyword.
Elemental abundances
Files with various elemental abundances are provided in the directory !xuvtop/abund/ Element abundances are in the usual dex notation (Log10 values, relative to H, that has a Log10 value of 12).
Options are available within the routines to choose different elemental abundances. User-defined abundance files can also be used, and should have a .abund file extension.
Be aware that any element missing in the elemental abundance file will also be missing in any output created by any software that reads the elemental abundance file.
There is a great deal of controversy over the variation of the elemental abundances in the solar and stellar atmospheres. See the reviews of Meyer (1985, 1993), Widing and Feldman (1992), Mason (1992), Raymond et al. (2001). Also, it should be kept in mind that different analyses can lead to very different results. For example, the ionisation balance, the selection of lines, and the spectroscopic method used can each account for a variation of a factor of two or more in the derived element abundances (see Del Zanna et al., 2002 and references therein).
Ionisation Fractions
Files giving collisional ionization equilibria are provided in the !xuvtop/ioneq directory. User defined ionisation files should have a .ioneq file extension.
Be aware that most CHIANTI software uses the temperatures in these files as a base for the calculations. For example, if DEM(T) values are supplied, they are first interpolated at the temperatures in the ionization fraction, and the calculations are done at those temperatures.
The ionisation fractions have been taken from the tabulated values in the published literature (e.g. Arnaud & Raymond, 1992; Arnaud & Rothenflug, 1985; Mazzotta et al., 1998).
Be aware that any ion missing in a ionisation fraction file will also be missing in any output created by the software.
Also, be aware that any line missing a temperature overlap with the chosen ionisation fraction would have zero emissivity and will not be output by the software.
Be aware that large differences between different tabulations are present, and that large uncertainties are associated with these calculations. It should be noted that the ionisation equilibrium plays a major role not only in the derivation of the DEM, but also in that one of the elemental abundances. In this respect, it is important to be aware of the fact that a number of ions, in particular those of the Li and Na isoelectronic sequence, present anomalous behaviour (see Del Zanna et al., 2002, and references therein).
Differential Emission Measure
Files specifying various standard differential emission measures (DEM) distributions for different solar features are provided in the !xuvtop/dem directory. Additional files for stellar atmospheres will also soon be added. Each file contains the Log10 T and Log10 DEM values in two columns, ordered with increasing temperature.
User-defined DEM files should have a .dem file extension and must have the same format and ordering of the files provided.
Be aware that any line missing a temperature overlap between the ion fraction and the chosen DEM distribution would have zero emissivity and will not be output by the software.
The emission measure distribution in the solar atmosphere is a complex issue. Starting with the pioneering work by Pottasch (1964), spectra in the UV wavelength range have been used to determine the distribution of material as a function of temperature, following various methods. More details can be found in Section 7.12.
Other files
Other files are in other miscellaneous directories. For example:
!xuvtop+'/ip/chianti.ip' has the ionization potentials for all the ions;
!xuvtop+'/continuum/ contains data used by the routines that calculate the continuum. For example, gffgu.dat contains the free-free gaunt factors of Sutherland (1998).
A number of Interactive Data Language (IDL) procedures are also provided as part of the CHIANTI package. These include routines to read the various CHIANTI database files, calculate level populations, line intensities, and temperature dependent and density dependent line intensity ratios.
Most of our efforts have gone into developing well-documented user-friendly IDL routines that meet readily apparent needs. We welcome contributions to the software.
CHIANTI has been run mainly on Sun, Dec Unix workstations and on PCs with Linux. CHIANTI also runs (with some small limitations) under Windows NT and in VMS. Please report to us any problems you might find.
All the IDL routines have been documented with extensive headers
giving detailed descriptions and
examples. Please read them carefully.
The CHIANTI routines can be grouped into three classes:
The CHIANTI routines are organised in a tree structure. The main level contains some high-level procedures and the HISTORY file, where all modifications to the software are logged.
Now, a description of the various high-level routines that are present within the CHIANTI software tree is given.
Compared to previous releases, significant changes occurred. Please read Sect. for details.
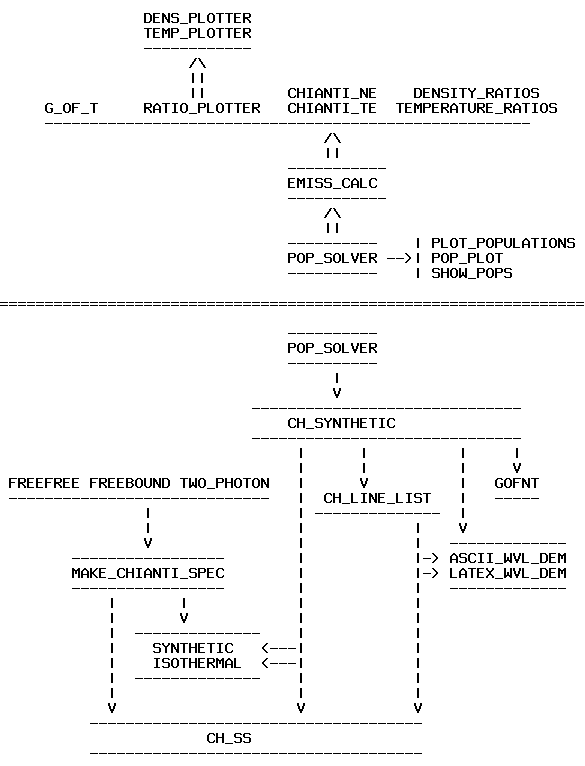
A new set of high-level and higher-level routines has been written for Version 4. The major change for V.4, aside from those already mentioned, has been the way to calculate synthetic spectra and handle line intensities (see Fig. 1):
Other routines that previously were only available within SolarSoft are also included now. The users therefore now have various different routines to choose from.
We have kept the older high-level routines, so the user can still use them as before (with slight modifications/additions of keywords). We have updated them and re-written as wrapper routines (essentially that call the newly-written routines) as follows:
| Synthetic spectra | ||
| ch_ss |
| |
| synthetic |
| |
| synthetic_plot |
| |
| isothermal |
| |
| make_chianti_spec |
| |
| Line intensities | ||
| ascii_wvl_dem |
| |
| latex_wvl_dem |
| |
| ch_synthetic |
| |
| ch_line_list |
| |
| Line emissivities | ||
| emiss_calc |
| |
| gofnt |
| |
| g_of_t |
| |
| Density-sensitive line ratios | ||
| dens_plotter |
| |
| density_ratios |
| |
| chianti_ne |
| |
| plot_chianti_ne |
| |
| Temperature-sensitive line ratios | ||
| temp_plotter |
| |
| temperature_ratios |
| |
| chianti_te |
| |
| plot_chianti_te |
| |
| Continuum | ||
| freefree |
| |
| freebound |
| |
| two_photon |
| |
| Level populations | ||
| plot_populations |
| |
| pop_plot |
| |
| show_pops |
| |
| Miscellaneous | ||
| rad_loss |
| |
| max_temp |
| |
| plot_ioneq |
| |
| chianti_dem |
| |
| plot_dem |
| |
| integral_calc |
| |
| ch_read_fits |
| |
| ch_write_fits |
| |
|
For the first two classes of routines, by simply typing the name of the routine, a description of how to call the routines, with examples, is printed. For example,
IDL > temperature_ratios IDL > temperature_ratios,ion,wmin,wmax,Log10(tempmin),Log10(tempmax),$ IDL > temperature,ratio,description,$ IDL > [pressure= ,density= , psfile= , outfile= ] IDL > IDL > i.e.: IDL > temperature_ratios,'c_5',40.,50.,5.,7.,temp,rat,desc
In any case the best way to understand what a routine does and how it works is to read the header documentation with e.g.:
IDL > xdoc,'ch_synthetic' IDL > doc_library,'ch_synthetic'
Another way to quickly see the keywords of a routine is to use:
IDL > chkarg,'temperature_ratios ..... ---> Call: pro temperature_ratios,ions,wmin,wmax,tempmin,tempmax,$ temperature,ratio,description,$ density=density,psfile=psfile, $ outfile=outfile,noprot=noprot, $ radtemp=radtemp,rphot=rphot,photons=photons, $ ioneq_file=ioneq_file, abund_file=abund_file, $ VERBOSE=VERBOSE
CHIANTI is currently distributed in two ways:
http://www.lmsal.com/solarsoft/
for details on how to download and install the package. The database and the software are organised in a self-contained package
$SSW/packages/chianti/
with the following tree structure:
dbase/ (database) doc/ (documentation, in particular the USER GUIDE) idl/ (IDL software) setup/ (supplementary setup files)
The contents of the SolarSoft CHIANTI package are
mirrored daily from a master tree.
Normally, only small fixes to the existing
software can occur rather frequently.
All modifications to the software are logged in the $SSW/packages/chianti/idl/HISTORY file.
Modifications to the database are much less frequent. They are described in the $SSW/packages/chianti/dbase/README_CHIANTI file.
We send an e-mail to the CHIANTI user group every time we make a minor release of the database available.
Note that the contents of the SolarSoft package change on a frequent timescale normally to fix bugs caused by the use of new IDL releases.
We recommend that you use CHIANTI within the SolarSoft framework and that you setup in your site a mirror in order to have automatic upgrades. It is easy to follow the instructions to download and setup the package.
Currently, the data and the software are distributed in two separate tar files. The tar files have a similar tree structure as the SolarSoft distribution.
E.g. the data are in CHIANTI_4.0_data.tar that contains a copy of $SSW/packages/chianti/dbase.
CHIANTI_4.0_pro.tar contains doc/, a copy of $SSW/packages/chianti/doc/ and idl/, a copy of $SSW/packages/chianti/idl/, plus idl/gen/, a copy of the $SSW/gen/ routines. This is because some routines of the $SSW/gen/ directory are needed to run some of the CHIANTI programs.
CHIANTI is a package, in the sense that database and progams are to be used together. The current version of the database must be used with the current version of the programs. Backward compatibility does not always apply.
To run any CHIANTI IDL procedure, the following is needed:
If you are using SolarSoft you should have the setup already organised so as to have the path of the CHIANTI IDL procedures added to IDL_PATH, the !xuvtop and the other IDL system variables defined. This is done automatically by using
unix> setssw chianti unix> sswidl or unix> sswidl IDL > ssw_packages,/chianti
After this, you will be able to run the CHIANTI routines.
Users of previous versions, please NOTICE:
The procedures to install CHIANTI have changed in small but important ways.
Download the CHIANTI files
Download the CHIANTI data tar file (e.g. CHIANTI_4.0_data.tar.gz) and the CHIANTI IDL procedures tar file (e.g. CHIANTI_4.0_pro.tar.gz) and put the tar files into a directory (for example, /data1/chianti/dbase for the data and /data1/chianti/ for the software) and then do the following:
unix> gunzip [file_name].tar.gz unix> tar xvf [file_name].tar
This will copy all the CHIANTI data files into /data1/chianti/dbase and create the /data1/chianti/idl and /data1/chianti/doc/ directories.
Define the IDL paths and the system variables
There are two ways of doing the above. The first is to define the system variables within IDL, the second is outside IDL. We suggest the first option. Once IDL is started, there are three steps:
unix > idl
Unix: IDL> !PATH = '+/data1/chianti/idl:'+!PATH Windows: IDL> !PATH = '+C:\data1\chianti\idl;'+!PATH VMS: IDL> !PATH = '+/data1/chianti/idl,'+!PATH
IDL> !PATH = EXPAND_PATH(!PATH)The '+' and the EXPAND_PATH are needed since the IDL routines are organised into subdirectories. The second option involves writing (UNIX) the following statement in your /.cshrc (or /.login) file:
setenv IDL_PATH /usr/local/rsi/idl_4/lib:+/data1/chianti/idl
(assuming you have the main IDL directory in /usr/local/rsi/idl_4).
Unix: IDL> use_chianti, '/data1/chianti/dbase' Windows: IDL> use_chianti, '+C:\data1\chianti\dbase'
After following the above steps, it will be possible to run the CHIANTI routines from any directory. use_chianti also allows you to set your default abundance and ionization equilibria files with the abund and ioneq keywords.
Previous CHIANTI users should check the note below.
We suggest that you add the three above calls to your IDL_STARTUP file (say /.idl_startup). If this file does not exist then it should be created. In UNIX, this can be done if you add the following line to your .login file:
setenv IDL_STARTUP ~/.idl_startup
(Note that the changes to the .login file mean that you should do a source ~/.login before running IDL).
Alternatively, you can write the three statements above in a file, say start_chianti.pro:
!PATH = '+/data1/chianti/idl:'+!PATH !PATH = EXPAND_PATH(!PATH) use_chianti, '/data1/chianti/dbase' ENDand run
IDL> .r start_chianti
Note to previous CHIANTI users:
If you had already defined the CHIANTI system variables
before entering IDL or in your IDL STARTUP file you
should remove those definitions.
Alternatively, instead of using use_chianti, '/data1/chianti/dbase', you have to make sure you have in your IDL STARTUP file something like this:
!PATH = '+/data1/chianti/idl:'+!PATH !PATH = EXPAND_PATH(!PATH) defsysv,'!xuvtop', '/data1/chianti/dbase' defsysv,'!ioneq_file','/data1/chianti/dbase/ioneq/mazzotta_etal.ioneq' defsysv,'!abund_file','/data1/chianti/dbase/abundance/cosmic.abund' defsysv,'!BCOLOR',0 defsysv,'!ASPECT',1.0
For a review on Spectroscopic Diagnostics in the EUV for Solar and Stellar Plasmas see e.g. Mason and Monsignori Fossi (1994).
The intensity I(lij), of an optically thin spectral line of wavelength lij (frequency nij = [ c/(h lij)]) is
| (1) |
where i, j are the lower and upper levels, Aji is the spontaneous transition probability, Nj is the number density of the upper level j of the emitting ion and h is the line of sight through the emitting plasma. In low density plasmas the collisional excitation processes are generally faster than ionization and recombination timescales, therefore the collisional excitation is dominant over ionization and recombination in populating the excited states. This allows the low-lying level populations to be treated separately from the ionization and recombination processes.
For allowed transitions we have Nj(X+m) Aji ~ Ne. The population of the level j can be expressed as:
| (2) |
See the routine PROTON_DENS described in Sect. B.5 for details on how to calculate N(H)/Ne.
In the `standard model' for interpreting line intensities there are three fundamental assumptions that serve to simplify the problem considerably:
The atomic data contained in the CHIANTI database are particularly suited to the analysis of emission lines via this model, and the following discussion outlines this approach. No attempt is made to discuss non-equilibrium conditions.
With the first of the assumptions, the
population of ions lying in a
given state is constant and so the number of ions leaving this state
per unit time must exactly balance the number arriving into that
state. If we denote the number of transitions leaving the state i to
a state j taking place per unit time per unit
volume by aij, then steady state implies
| (3) |
| (4) |
| (5) |
Now our second assumption means that the processes that
affect the ionisation state of the plasma do not affect the quantity
ni. Eq. 5 thus becomes
| (6) |
For the basic CHIANTI model these processes are simply electron and proton excitation and de-excitation, and the generalised radiative decay:
| (7) |
Cije is given by:
| (8) |
| (9) |
where wi is the statistical weight of level i, k is
Boltzmann's constant, T the electron temperature, and qij
the electron excitation rate coefficient which is given by:
| (10) |
The solution of Eq. 6 is performed by the CHIANTI routine pop_solver.pro, which gives the fraction of ions in the state i.
The level populations for a given ion can be calculated and displayed with plot_populations.pro (but also see pop_plot.pro).
We rewrite the intensity as:
| (11) |
where the function
| (12) |
called the contribution function, contains all of the relevant atomic physics parameters and is strongly peaked in temperature.
gofnt.pro calculates these contribution functions (see also g_of_t.pro for a slightly different way of calculating contribution functions).
Please note that in the literature there are various definitions of contribution functions. Aside from having values in in either photons or ergs, sometime the factor [ 1/(4p)] is not included. Sometimes a value of 0.83 for N(H)/Ne is assumed and included. Sometimes the element abundance factor is also included. Any of the above (or any other) variations also affect the definition of a line intensity in terms of the contribution function and the DEM. In the following we will refer to the functions C(T,lij,Ne) and G(T,lij,Ab(X),Ne) = Ab(X) C(T,lij,Ne) ( i.e. the contribution function that contains the abundance factor ).
If we define, assuming that is a single-value function of the temperature, the differential emission measure DEM (T) function as
| (13) |
the intensity can be rewritten, assuming that the abundance is constant along the line of sight:
| (14) |
The DEM gives an indication of the amount of plasma along the line of sight that is emitting the radiation observed and has a temperature between T and T+dT.
The IDL routine chianti_dem.pro described in Sect. 7.12 calculates the Differential Emission Measure DEM(T) using the CHIANTI database, from a given set of observed lines.
Routines such as ch_synthetic.pro (see Sect. 7.1 and Sect. E.1) calculate line intensities without the abundance factor, that is only included at a later stage.
In the isothermal approximation, all plasma is assumed to be at a single temperature (To) and the intensity becomes:
| (15) |
where we have defined the column emission measure
| (16) |
ch_synthetic.pro in the isothermal approximation calculates I = C(To, lij, Ne) rNe NH dh, while isothermal.pro and ch_ss.pro (see examples in Sect. 7.1) can be used to create synthetic spectra (with the abundance factor).
It is also possible to calculate intensities and spectra with a multi-temperature model, by providing an array of To, EMh values.
Please note that in the literature many different definitions of Differential Emission Measures, Emission Measures and approximations can be found (see Del Zanna et al., 2002 for some clarifications).
In the stellar case, the theoretical flux of an optically thin spectral line is:
| (17) |
where C(Ne, T, lij) has the
same expression as above, d is the star's distance,
dV is the volume element, and V is the entire source volume.
A volume Differential Emission Measures DEM is often defined:
| (18) |
| (19) |
At the moment CHIANTI does not include volume emission measures. In the near future we will modify the software and the definition of the DEM in order to include volume emission measures.
However, any volume Differential Emission Measures can be rescaled to column DEMs and used within the software to produce synthetic spectra for stellar coronae. One way of doing this is to assume spherical symmetry, and that the emitting region is a layer dh distributed over the entire star's disk, i.e. dV=4pR2* dh (R* is the star's radius). If the star's radius and distance are known, a volume DEM can be scaled with the factor [(4pR2*)/(d2)] to obtain a column DEM.
If this is used, the outputs will have flux units, i.e. ergs cm-2 s-1 (or photons cm-2 s-1) and not ergs cm-2 s-1 sr-1.
An example of scaled DEM is provided in the file AU_Mic.dem, in the CHIANTI distribution.
Column DEMs and EMs are assumed when the spectra are folded with effective areas (see Sect. 7.1). The effective areas are assumed to have units of counts photons-1 cm+2, so the output units of the spectra will be counts s-1 pixel-1.
Also note that corrections to interstellar absorption are not presently included in CHIANTI.
For each ion for which proton rates are available, an additional file is required in the database to contain the fits to the rate coefficients. The file has the suffix .PSPLUPS, and is exactly analogous to the .SPLUPS file for the electron fits. All of the proton transitions included in CHIANTI are forbidden transitions taking place between levels within the same configuration. Many of the transitions required 9-point splines (see Sect. C) in order to provide adequate fits. The number density of protons, Np, is calculated with the IDL routine proton_dens.pro (see Sect. B.5).
By default, all routines will include proton rates in the calculation of the ion level balance. A keyword /NOPROT can be used to switch off the proton rates.
Within CHIANTI, we presently model the Photoexcitation and Stimulated Emission by assuming a a blackbody radiation field of temperature T*. The generalized photon rate coefficient in this case is:
| (20) |
where Aji is the radiative decay rate and W(R) is the radiation dilution factor which accounts for the weakening of the radiation field at distances R from the source center.
We also assume an uniform (no limb brightening/darkening) spherical source with radius R*:
| (21) |
where
| (22) |
It is important to remember the assumptions in our formalism for radiation processes. For a given ion, only very specific wavelengths in the radiation continuum will affect the ion's level balance. If there are significant deviations from a blackbody spectrum at any of these wavelengths (perhaps due to a deep absorption line) then CHIANTI does not model the ion entirely correctly.
Examples of specific uses of the extra radiation processes include modeling of coronal emission lines above the surface of the Sun and other cool stars when the coronal electron density falls to low enough values that electron collisions lose their potency.
For the Sun, photoexcitation is very important for the infrared coronal lines. Photoexcitation is also important for modelling nebular ions that are irradiated by a hot star, such as in planetary nebulae, symbiotic stars and Wolf-Rayet stars.
No additions or modifications to CHIANTI data files are required for photoexcitation and stimulated emission as their rates are entirely determined from the radiative decay rates, level separation energies, and statistical weights - information already contained in CHIANTI. It is only necessary to specify the radiation field temperature and the dilution factor. These are specified as inputs to the IDL procedures through the new keywords RPHOT and RADTEMP. RPHOT specifies r = [ R/(R*)], while RADTEMP gives the blackbody radiation temperature in K.
By default, photoexcitation and stimulated emission are not included in the level balance equations unless the keywords are set.
An IDL routine to include the two photon continuum has been added to CHIANTI, while the free-bound and free-free continuum (bremsstrahlung) routines have been revised. See Young et. al. (2002) for more details.
Note that the output units of the continuum routines are by default 10-40 ergs sr-1 s-1 Å-1 per unit emission measure rNe NH dh.
On the other hand, the SolarSoft routine CONFLX outputs a continuum in photons s-1 Å-1 assuming an emission measure rNe2 dh = 1050.
The two-photon continuum is calculated with two_photon.pro.
Transitions in hydrogen-sequence ions
The first excited level (2s 2S1/2) of the hydrogen iso-electronic sequence ions can decay only by means of forbidden magnetic dipole and two-photon transitions. The importance of the competing magnetic dipole transition increases with Z but for nickel (Z=28), the two-photon transition rate is roughly 5 times that of the magnetic dipole rate.
The spectral emissivity (erg cm-3 s-1 sr-1 Å-1) for optically-thin two-photon emission at wavelength l is given by:
| (23) |
where Aj,i (sec-1) is the Einstein spontaneous emission coefficient (A value); Nj(X+m) is the number density of the level j of the ion X+m; f is the spectral distribution function; and l0 is the wavelength corresponding to the energy difference between the excited and ground level.
Two-photon continuum transitions in helium-sequence ions
For the helium iso-electronic sequence, the second excited level (1s2s 1S0) decays through a forbidden magnetic dipole and two-photon transitions.
The bremsstrahlung emission is calculated with freefree.pro. This routine has been rewritten ex-novo. It now includes the Itoh et al. (2000) and Sutherland (1998) gaunt factors.
Itoh et al. (2000) have provided an analytical fitting formula for the relativistic thermal bremsstrahlung gaunt factors, and this is now added to CHIANTI. The fitting formula is valid for the ranges 6.0 # log T # 8.5 and -4.0 # log (hc/klT) # 1.0. For temperatures below log T=6.0 we retain the non-relativistic Gaunt factors of Sutherland (1998) for computing the continuum. The condition log (hc/klT) # 1.0 results in some of the low wavelength points being inaccurately represented by the Itoh et al. fitting formula. For these wavelengths the Gaunt factors of Sutherland (1998) are used to compute the continuum level.
The relativistic free-free continuum is almost identical to the non-relativistic continuum at low temperatures. At T=1×108 K (the maximum temperature permitted by the ion balance calculations contained in CHIANTI) the relativistic continuum is around 1% higher near the peak of the distribution.
The free-bound continuum emission is calculated with freebound.pro. This routine has been rewritten. The new routine uses the the Karzas and Latter (1961) approximation to the photoionization cross-sections and calculates free-bound gaunt factors for levels n=1-6. Additional data files have been created for this purpose. For example, free-bound radiation produced by recombination of an electron onto C IV to produce C III will use the data in the c\_3.fblvl file.
In what follows we review the main points about the new software. We hope you find it useful and enjoy using it !
For an user-friendly, widget-based approach the best option is to use CH_SS:
IDL >ch_ss
This widget allows the user to calculate synthetic spectra in two basic steps. Basically, you follow the various widgets from top left to lower right to set the desired parameters. First calculate the line intensities. These values can be saved for later use. Next, specify further parameters such as the elemental abundances and instrumental spectral resolution and then calculate and plot the spectrum. These values can also be saved for later use. The HELP buttons in the widget provide short descriptions of the required information. More details are given below.
Alternatively, for e.g. background jobs, the
routine CH_SYNTHETIC can be used.
ch_synthetic.pro
calculates line intensities assuming
constant pressure or density (or a model T,N),
without the abundance factor.
One of the reasons why element abundances are not
included in the line intensities calculation is so that it is easier
for the user to see how modifying abundances affects their spectra in
e.g. ch_ss.pro.
The calling sequence is:
IDL> ch_synthetic, wmin, wmax, output=output, err_msg=err_msg, msg=msg, $
pressure=pressure, density=density, $
model_file=model_file, all=all,sngl_ion=sngl_ion, $
photons=photons, masterlist=masterlist, $
save_file=save_file , verbose=verbose,$
logt_isothermal=logt_isothermal, logem_isothermal=logem_isothermal,$
goft=goft, ioneq_name=ioneq_name, dem_name=dem_name, $
noprot=noprot, rphot=rphot, radtemp=radtemp, progress=progress
The routine has many KEYWORDS (see Sect. E.1 for a full list) A series of parameters must be set:
Wmin, Wmax: minimum maximum of the desired wavelength range in Angstroms
Pressure: pressure in emitting region (Pe, cm^-3 K).
Only a single value is accepted, and the calculation is
performed at constant pressure.
Density: density in emitting region (Ne, cm^-3).
Only a single value is accepted, and the calculation is
performed at constant density, unless LOGT_ISOTHERMAL is
defined. In this case, DENSITY can be an array of values, but
has to have the same number of elements as LOGT_ISOTHERMAL.
model_file Full path of the (Te,Ne) file if defined.
This file should have two columns, one with the Te (K)
values, and one with the Ne (cm^-3) values. If these
values are not sorted in ascending order of Te, the
routine does sort them.
The line intensities are calculated either in the isothermal approximation, in which case the following has to be defined:
LOGT_ISOTHERMAL: Array of logarithmic temperatures. LOGEM_ISOTHERMAL: Array of logarithmic emission measures (0 by default).
or by folding the G(T) with a differential emission measure DEM contained in the file specified by DEM_NAME. The program will prompt the user to select one if not defined.
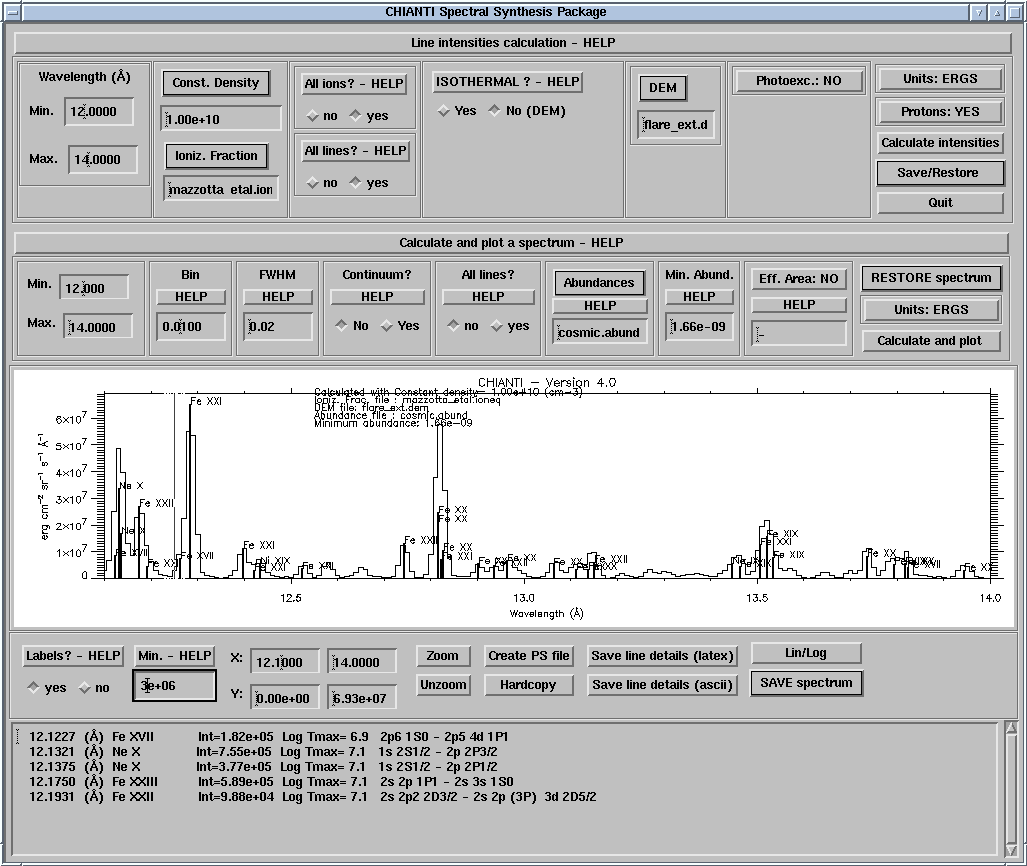
Example:
IDL> ch_synthetic, 10,20., output=str , pressure=1.e+15,$
ioneq_name=concat_dir(concat_dir(!xuvtop,'ioneq'),'mazzotta_etal.ioneq'),$
dem_name=concat_dir(concat_dir(!xuvtop,'dem'),'flare.dem'),$
/photons, /noprot, /all, sngl_ion=['fe_17','fe_18']
Creates an output structure str that contains the line intensities of only Fe XVII and Fe XVIII in the 10-20 Å range calculated at constant pressure of 1015, with the ionization balance in mazzotta_etal.ioneq and the DEM values in flare.dem in the standard CHIANTI distribution (if not supplied these files can be selected with a widget). Line intensities are in photons cm-2 s-1 sr-1 (KEYWORD photons), the proton rates are not included (KEYWORD noprot), and all the lines in the database (KEYWORD all) are included (also the lines with only theoretical energy levels).
You can see the contents of the structure with e.g.
IDL> help, str,/st IDL> help, str.lines[0],/stThe last command shows the first structure associated with the first spectral line.
The CHIANTI line intensities structure can be saved and later restored from the command line in various ways. We suggest two:
IDL> savegen, file='ch_int_10_20_fe.genx', struct=str IDL> restgen, file='ch_int_10_20_fe.genx', struct=str
to save and restore the IDL structure str in the file ch_int_10_20_fe.genx.
Please note that we discourage the use of e.g.:
IDL> save, file='output.save', output IDL> restore, file='output.save'since IDL save files generated with later versions of IDL are usually not readable with earlier versions.
IDL> ch_write_fits, str, 'output.fits' IDL> ch_read_fits,'output.fits', str
to save and restore the IDL structure str in the FITS file output.fits. Aside from an introductory HEADER, the contents of the IDL structure are converted into two binary tables. Extensive comments are added.
In either case, the structure saved in the .genx and .fits files can be restored via CH_SS to later create a spectrum.
For an user-friendly approach the best option is to use CH_SS:
IDL >ch_ss
Alternatively, if you have already calculated a line intensity structure (as shown above), you can use CH_LINE_LIST. This program creates a latex or an ascii file of predicted spectral line intensities and wavelengths corresponding to selected parameters.
The routine has many KEYWORDS. Please read the header or read Sect. E.3 for details. The calling sequence is:
IDL> ch_line_list, transitions, outname, latex=latex, ascii=ascii, $
wmin=wmin,wmax=wmax,$
SPECTRUM=SPECTRUM, abundfile=abundfile, min_abund=min_abund, $
minI=minI,photons=photons,kev=kev, $
all=all,no_sort=no_sort, sngl_ion=sngl_ion
Example:
IDL> restgen, file='ch_int_10_20_fe.genx', struct=tran
IDL> ch_line_list, tran, 'ch_line_list.tex', /latex,$
abundfile=concat_dir(concat_dir(!xuvtop,'abundance'),'cosmic.abund'),$
mini=1e13
This creates a latex file ch_line_list.tex where only lines with an intensity greater than 1013 (KEYWORD mini) are included, and the allen.abund file in the standard CHIANTI distribution is used (if not supplied it can be selected with a widget).
Then, you have to latex the file three times, and optionally xdvi it:
unix> latex ch_line_list unix> latex ch_line_list unix> latex ch_line_list unix> xdvi ch_line_list
If you do not have it already, you will need the package longtable.sty that is distributed as part of ftp://cam.ctan.org/tex-archive/macros/latex/required/tools.tar.gz
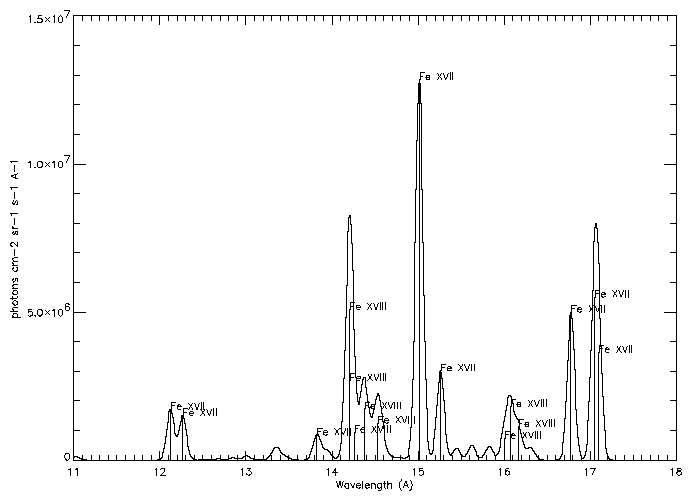
You will obtain a table that looks like:
|
| ||||
| Ion | l (Å) | Transition | Tmax | Int |
| Fe XVII | 12.1227 | 2p6 1S0 - 2p5 4d 1P1 | 6.9 | 1.11e+14 |
| Fe XVII | 12.2639 | 2p6 1S0 - 2p5 4d 3D1 | 6.9 | 9.81e+13 |
| Fe XVII | 13.8231 | 2p6 1S0 - 2s 2p6 3p 1P1 | 6.9 | 6.25e+13 |
| Fe XVIII | 13.9540 | 2s2 2p5 2P3/2 - 2p4 (1S) 3d 2D5/2 | 6.9 | 2.06e+13 |
| Fe XVIII | 14.1519 | 2s2 2p5 2P3/2 - 2p4 (1D) 3d 2D3/2 | 6.9 | 1.35e+13 |
Alternatively, you can also create a latex file with a list of line identifications and intensities using the wrapper routine LATEX_WVL_DEM:
IDL > latex_wvl_dem,100.,200., pressure=1.e+15,mini=1.However, latex_wvl_dem calls ch_synthetic and ch_line_list, and if you want to modify some of the parameters of ch_line_list (e.g. the minimum intensity) you will have to redo the calculation which will take some time. Windows will pop up so that you can select the abundance, the ionization equilibrium and the differential emission measure files. This will create by default a file linelist.tex in the user's working directory, by default.
To create an ascii file with the line details you can follow a similar approach, i.e.:
IDL> restgen, file='ch_int_10_20_fe.genx', struct=tran
IDL> ch_line_list, tran, 'ch_line_list.ascii', /ascii,$
abundfile=concat_dir(concat_dir(!xuvtop,'abundance'),'allen.abund'),$
mini=1e13
Alternatively, you can also use the wrapper routine
IDL > ascii_wvl_dem,100.,200.,pressure=1.e+15,mini=1.However, ascii_wvl_dem calls ch_synthetic and ch_line_list, and if you want to modify some of the parameters of ch_line_list (e.g. the minimum intensity) you will have to redo the calculation which will take some time.
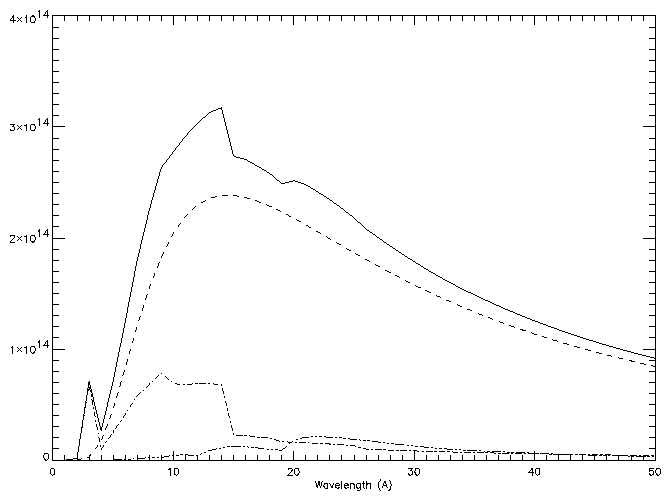
For example, to calculate the free-free, free-bound and two-photon continuum at a temperature of 5 × 106 K, for wavelengths at 1 Å intervals between 1 and 50 Å:
freefree,5.e+6,findgen(50)+1.,ff freebound,5.e+6,findgen(50)+1.,fb two_photon,5.e+6,findgen(50)+1.,tp window,0 plot,findgen(50)+1.,ff+fb+tp,xtit='Wavelength (A)' oplot, findgen(50)+1.,ff,line=2 oplot, findgen(50)+1.,fb,line=3 oplot, findgen(50)+1.,tp,line=4
|
Note that the intensities are in units of 10-40 ergs cm3 s-1 sr-1 Å-1 per unit emission measure rNH Ne dh (cm-5).
If DEM values are passed to the routines (via the keyword DEM_INT), it is assumed that they are given as NH Ne dh/dT . The units are 10-40 ergs cm-2 s-1 sr-1 Å-1 in this case.
The structure created by CH_SYNTHETIC can be restored via CH_SS to create a spectrum. Alternatively, it can be used as an input to the program MAKE_CHIANTI_SPEC. This program creates the CHIANTI SPECTRUM structure (read Sect. E.5 for details), an OUTPUT structure similar to the structure created by CH_SYNTHETIC, with some additional tags. The calling sequence is:
IDL> make_chianti_spec, TRANSITIONS, LAMBDA, OUTPUT, BIN_SIZE=BIN_SIZE, $
INSTR_FWHM=INSTR_FWHM, BINSIZE=BINSIZE, $
WRANGE=WRANGE, ALL=ALL, continuum=continuum, $
ABUND_NAME=ABUND_NAME, MIN_ABUND=MIN_ABUND, $
photons=photons, file_effarea=file_effarea, $
err_msg=err_msg, verbose=verbose
The routine has many keywords and options. Please read Sect. E.4 for details.
IDL> restgen, file='ch_int_10_20_fe.genx', struct=tran
IDL> make_chianti_spec, tran, lambda, struct,/CONTINUUM, $
BIN_SIZE=0.01, instr_fwhm=0.1, WRANGE=[10.,19.],$
abund_name=concat_dir(concat_dir(!xuvtop,'abundance'),'cosmic.abund')
|
Some Caveats:
You may find that the calculation is slow. This is usually due to the continuum calculation. In general, it is advisable not to calculate spectra over large wavelength ranges. In any case you can speed up the continuum calculation by reducing the numbers of elements, using the KEYWORD MIN_ABUND.
To see the contents of the structure:
IDL> help, struct,/st IDL> help, struct.lines[0],/st
While to show the spectrum and the main contributing lines:
IDL> window,0 & plot,struct.lambda,struct.spectrum
for i=0,n_elements(struct.lines) -1 do $
if struct.lines[i].peak gt 7e5 then $
xyouts, struct.lines[i].wvl, struct.lines[i].peak, struct.lines[i].snote
It may be useful to save the SPECTRUM structure, that can be later inspected with the widget CH_SS:
IDL> savegen, file='ch_spectrum_10_20_fe.genx', struct=struct IDL> ch_write_fits, struct, 'ch_spectrum_10_20_fe.fits'
Alternatively, the wrapper routine SYNTHETIC (see Fig 1) can also be used to calculate CHIANTI line intensities. For example:
IDL > synthetic, 150., 200., 1., pressure=1.e+15, wvl, spectrum, list_wvl, list_ident
will create a synthetic spectrum with a resolution of 1 Å between 150 and 200 Å for a specified set of abundances and differential emission measure at a constant pressure of 1.e+15 (Ne T cm-3 K). The output arrays wvl, spectrum contain the wavelengths and the intensities (in erg cm-2 s-1 sr-1 Å-1 by default). The output arrays ist_wvl, list_ident contain the list of wavelengths and descriptions of the lines that made up the spectrum.
Windows will pop up so that the user can select the abundance file, the ionization equilibrium and the differential emission measure. A spectrum is created by convolving with a Gaussian profile with a FWHM of 1 Å. If the /CONTINUUM keyword had been set, then the continuum would also have been calculated and added to the spectrum. To plot the spectrum and interactively identify lines:
IDL > synthetic_plot, wvl, spectrum, list_wvl, list_ident, 2.
by clicking the left mouse button, a list of predicted lines within 2 Å of the selected wavelength will be printed out along with their predicted intensity. Clicking the right mouse button will exit the procedure.
For an user-friendly approach the best option is to use CH_SS:
IDL >ch_ss
Alternatively:
IDL > isothermal, 150., 200., 1., [1.e6], wvl, spectrum,$
list_wvl, list_ident, edensity=1.e9,$
ioneq_name=!xuvtop+'/ioneq/mazzotta_etal.ioneq',$
abund_name=!xuvtop+'/abundance/cosmic.abund'
IDL> synthetic_plot, wvl, spectrum, list_wvl, list_ident, 1.
calculates an isothermal synthetic spectrum with a resolution of 1 Å between 100 and 200 Å for a specified set of abundances and differential emission measure at a constant density Ne = 109 cm-3. The output arrays wvl, spectrum contain the wavelengths and the intensities (in erg cm-2 s-1 sr-1 Å-1 by default). The output arrays ist_wvl, list_ident contain the list of wavelengths and descriptions of the lines that made up the spectrum. synthetic_plot can then be used to view the spectrum.
Note: isothermal now is a wrapper routine that calls ch_synthetic . It has particular features. Please read the header documentation.
|
CH_SS is an user-friendly multi-purpose (see Fig 1) widget that allows the calculation of line intensities (calling CH_SYNTHETIC) and of a synthetic spectrum (calling MAKE_CHIANTI_SPEC) by merging line intensities and continua. The parameters can be interactively set, and the results visually inspected. Line intensities can be saved and restored in various ways. The results can also be stored in various ways, ranging from output plots to tables of line details (using CH_LINE_LIST) or save files.
CH_SS replaces the CHIANTI_SS procedure. The calling sequence is:
IDL> ch_ss, font=font
Note that if the widget appears too large you can change the font. The widget is organised into four Sections:
This can be done in two ways:
1-Restore a save file with the CHIANTI line intensities already calculated. This is done with the RESTORE button. .genx and .fits files can be restored.
2-Calculate CHIANTI line intensities with a call to CH_SYNTHETIC.
In this case, A series of parameters must be set:
If set to no, then it is possible to select a list of ions with a widget
If set to yes, also the lines that do not have corresponding observed energy levels are included. In this case, the wavelengths are calculated from the theoretical energy levels, and might not be very accurate.
If set to yes, then the user is requested to enter one or more temperatures (as logarithmic values - Log T ) and correspondent column emission measures EM logarithmic values. NOTE: if more than one value is entered, then the sequence must be separated by commas (e.g.: 6.0, 6.5, 7.), and both Log T and Log EM must have the same number of values
Once all the parameters have been defined, the user should click on the "Calculate intensities" button to start the calculation (which calls CH_SYNTHETIC).
Once the calculation is finished, an IDL structure is loaded into memory. It is then possible to save it for later use by clicking on the "SAVE" button.
Once the IDL structure with the line intensities is in the memory, it is then possible to calculate and plot a spectrum (SECTION 2).
This section controls the parameters that are needed to fold the line intensities and the continua into a synthetic spectrum. These parameters are used by MAKE_CHIANTI_SPEC.
Before this is done, a set of line intensities MUST be in the program memory. This is done either by calculating the intensities or by restoring a save file with previously calculated values (SECTION 1). Setting the parameters:
Also note that to have the correct output units (counts s-1 bin-1) the appropriately scaled DEM (or EM) values must be provided.
After this, by clicking on the "Calculate and plot" button the program calculates and plots the synthetic spectrum.
Once the spectrum is displayed, it is then possible to view the details of the lines by clicking with the mouse in the plot window, and to perform various operations by clicking on the buttons in SECTION 3
This Section allows the user to select a few parameters for the plotting, and to create different types of OUTPUT.
NOTE that only the line details and portion of the spectrum shown will be output.
Finally, SECTION 4 is a text information window, where various messages are printed.
Clicking the cursor on any part of the displayed spectrum will give a listing of the lines within a range of Angtroms of that wavelength. Text information on the lines is printed.
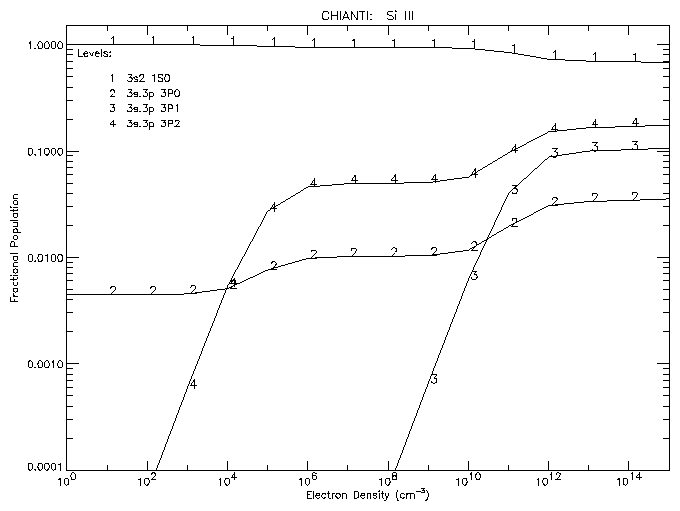
To plot the populations of the first 4 levels of Si III as a function of density at a temperature of 3 x 104 K:
IDL > plot_populations,'si_3',3.e+4,4
|
Optionally, output files can be created.
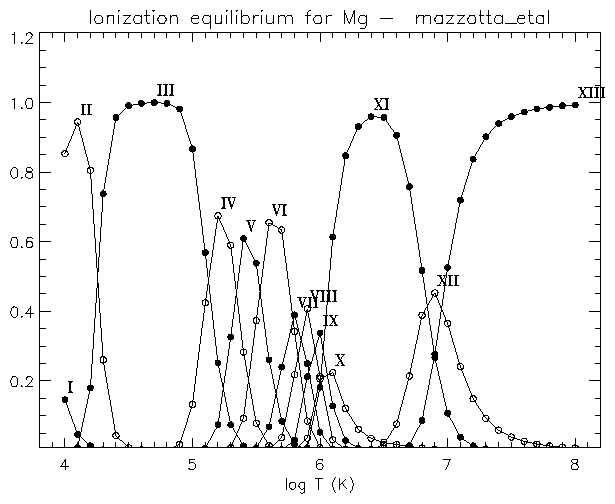
If you are interested to see the differences between the various ionisation equilibria for e.g. Mg, you can use:
IDL > plot_ioneq,'Mg'
You will be able to select one of the files, and optionally create a postscript file of the plot.
|
If you are only interested in e.g. the Mg VIII, Mg IX, Mg X ions, you can type:
IDL > plot_ioneq,'Mg', ion=[8,10]
If, instead, you are interested in obtaining the temperature at the maximum ionisation fraction for e.g. Mg X, you can use:
IDL > print, max_temp ('Mg X')
You will be asked to select an ionisation equilibrium file.
Spectroscopic diagnostic line ratios in the UV wavelength range have been used extensively to determine the electron density and temperature in the solar atmosphere (cf Dere and Mason, 1981, Gabriel and Mason, 1982, Mason, 1991, Mason and Monsignori Fossi, 1994). The theoretical intensity ratios from individual ion species provide a measurement of electron density which is independent of any assumptions about the volume of the emitting region. This is of particular importance in the transition region and coronal structures. The electron density (which determines the electron pressure) is an essential parameter in the study of energy transfer mechanisms. The routines that can be used are described below.
DENS_PLOTTER and TEMP_PLOTTER are high-level widgets for the analysis of density- and temperature-sensitive ratios of lines from the same ion. They allow inclusion of proton rates and photoexcitation. The calling sequence is simple:
IDL > dens_plotter,'o_5'
to study O V.
IDL > temp_plotter,'c_4'
to study C IV.
Alternatively, you can use the command-line routines, DENSITY_RATIOS and TEMPERATURE_RATIOS. They also allow inclusion of proton rates and photoexcitation via KEYWORDS.
The routine DENSITY_RATIOS plots the variation of line intensities with electron density, allowing density diagnostics to be studied. As an example, we can look for density sensitive line ratios of O V in the 1000 to 1500 Å wavelength region for densities between 108 and 1013 cm-3:
IDL > density_ratios,'o_5',1000.,1500.,8.,13.,den,rat,desc
two windows will open and plot the relative intensities of a few O V lines. To choose the ratio of 1371.294 to 1218.393 Å line, select first the 1371.294 Å line. Another widget will appear to select the denominator. Select the 1218.393 Å line. This will chose the ratio of 1371.294 to 1218.393 which will be plotted in a new window. Values of the density and intensity ratio will be put into the variables den and rat and desc will contain a descriptive string.
IDL > print, desc IDL > CHIANTI V. 4.0 O V 1371.2939 (E)/1218.3929 (E) T = 2.51e+05 (K)
The DENSITY_RATIOS procedure also allows to calculate the ratio at user-defined value of constant temperature. Blends are accounted for via a selection of lines.
To calculate temperature sensitive line ratios of C IV for lines between 100 and 1600 Å for temperatures between 104 and 106 K:
IDL > temperature_ratios,'c_4',100.,1600.,4.,6.,temp,rat,desc
As with density_ratios, a widget will appear that will allow you to select the numerator. Select the 384.175 and 384.190 Å lines as these will typically be blended in most spectrographs. Select the 1550.775 Å line for the denominator. The ratio of (384.175 + 384.190 Å) to the 1550.775 Å line as a function of temperature will be plotted and stored in the variables rat and temp, respectively. The TEMPERATURE_RATIOS procedure also allows to calculate the ratio at user-defined values of either constant pressure or constant density.
IDL > print, desc IDL > CHIANTI V. 4.0 C IV 384.1750+384.1900 (E)/1550.7750 (E) Ne = 1.00e+10 (cm!e-3!n)
The IDL procedure which calculates line intensity ratios as a function of electron density is called CHIANTI_NE. Another IDL procedure to calculate ratios sensitive to electron temperature is called CHIANTI_TE. For example,
IDL > chianti_te
Or
IDL > chianti_ne
User interactions with the main widget setup the wavelength range and other parameters that are described individually below. On the left hand side are controls for wavelength and electron density or temperature selection and on the right hand side for ion selection.
Ion selection
Select first the element and then the ion stage using the pull-down menus. Only those elements and ions currently available in the CHIANTI database are displayed for selection.
Wavelength ranges
There are up to 4 wavelength ranges that can be defined in order to restrict the calculation (and the number of lines). At least one should be defined. Default is to calculate all lines from 1 to 1700 Å.
Electron Density/Temperature Range
For CHIANTI_NE, select the electron number density range (Log10Ne) over which the intensity ratios are calculated. Default values are 106 to 1014 cm-3. The intensity ratios are calculated at constant temperature corresponding to the peak ionic abundance Tmax, unless the value of the temperature is typed in.
For CHIANTI_TE, select the electron temperature (Log10Te) over which the intensity ratios are calculated. Default values are 104 to 108 K. The intensity ratios are calculated at a constant electron number density corresponding to 108 cm-3, unless a different value is typed in.
minimum intensity ratio
The relative intensity of all the lines found in the wavelength range is calculated. Only the lines that have an intensity (relative to the brighter one) greater than 0.0001 (by default) are selected and displayed.
Units for the ratio plot
The ratios can either be calulated from the relative intensities in ergs (default) or photons.
Controlling the procedure
The action of both CHIANTI_NE and CHIANTI_TE is controlled via the buttons in the central panel of the display. From left to right these are:
QUIT - click on this to exit from the program, all plot windows are also deleted.
CALCULATE LINE INTENSITIES - using the wavelength ranges as defined in the widgets above. Two plots will apear in the window - on the left the Ni*A/Ne and on the right the intensity ratios, where Ni is the level population and A is the radiative transition probability (s-1). A list of spectral lines in the given wavelength range for that ion is displayed in the message window. The reference index is in the first column, then the wavelength, intensity and transition.
PLOT RATIO - prompts for and then plots specific line intensity ratios, using line indices available from the list. The ratios within a particular ion can be stored for later use using the SAVE button. To allow for blended lines in the observed spectra, multiple line indices can be given for the numerator and denominator. The format is fairly flexible but the nominator and denominator specification must be separated by a '/'. Otherwise the line indices can be separated by spaces or commas. The ratio values can be plotted either with a linear or a log scale.
HARDCOPY - the menu under this button will allow a variety of hardcopy plots (ratio plots or intensity plots) and the line details (+refs), which gives a record of the input. A line ratio has to be defined.
SAVE - it is sometimes useful to save the plotted line intensity ratios to study several different ions. You will be asked to enter file name to store ratio data. A line ratio has to be defined.
In the CHIANTI_NE case, a .CH_NE will be appended. An IDL structure called NE_RATIO will be saved. It has the TAGS: DENSITY,RATIO, TEMPERATURE, UNITS, COMMENT, DESC. The first two have the arrays of the densities and ratios, TEMPERATURE has the value of the constant temperature used, while the last three have a description of the units used, how the calculation was performed, and a description of the lines used to defined the ratio.
In the CHIANTI_TE case, a .CH_TE will be appended. An IDL structure called TE_RATIO will be saved. It has the TAGS: TEMP, RATIO, DENSITY, UNITS, COMMENT, DESC. The first two have the arrays of the temperatures and ratios, DENSITY has the value of the constant density used, while the last three have a description of the units used, how the calculation was performed, and a description of the lines used to defined the ratio.
Please note that previous versions of the above routines did not create the TEMPERATURE (DENSITY), UNITS, COMMENT tags.
The save file can be restored and replotted outside of CHIANTI_NE and CHIANTI_TE using the procedures PLOT_CHIANTI_NE and PLOT_CHIANTI_TE respectively, or used within user-defined procedures.
DELETE PLOT WINDOWS - to clear the ratio plots from the screen.
The ratio values stored in the save files created with the CHIANTI_NE and CHIANTI_TE routines can be replotted outside of CHIANTI_NE and CHIANTI_TE using the procedures PLOT_CHIANTI_NE and PLOT_CHIANTI_TE respectively. For example,
IDL > plot_chianti_te
Or
IDL > plot_chianti_ne
If no files are specified, a widget allows you to select any '*.CH_NE' or '*.CH_TE' files.
A LOG keyword allows the user to have the plot in a log scale.
It is also now possible to interactively modify the X and Y ranges and to create a postscript file. Users can modify these routines to e.g. over-plot observed data points.
Note: If you are interested in determining an isothermal temperature by using the ratio of lines emitted by different ions (and/or elements), then a possible way is to first calculte the contribution functions of the lines you are interested, and then calculate their ratio. Note, however, that such determinations can be very inaccurate, since they depend on the ionisation equilibrium chosen (and eventually on the element abundance).
To calculate the contribution function ( erg cm3 s-1 sr-1 by default) vs. temperature at a specified abundance, ionization equilibrium and pressure or density for the Fe XXIV line at 255.1 Å:
IDL > gofnt,'fe_10',170.,180.,temperature,g,desc
temperature, g are the arrays with the temperatures and the G(T) values. It is possible to calculate the G(T) at either constant electron density or pressure, via the KEYWORDS DENSITY or PRESSURE.
The KEYWORDS ABUND_NAME, IONEQ_NAME allow to run the routine in the background, giving names of the abundance and ionization fractions files.
The routine GOFNT allows the user to select a number of lines. If this is done, then the total sum of the G(T)'s of the selected lines is returned and plotted.
Optional outputs can be created. The default units are erg cm3 s-1 sr-1, unless the KEYWORD /PHOTONS is set, in which case the units are photons cm3 s-1 sr-1.
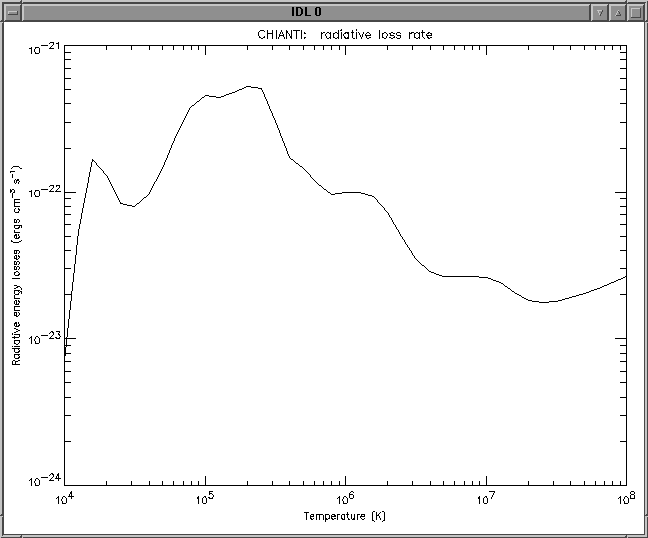
A procedure ('RAD_LOSS') calculates the total radiative loss rate as a function of temperature for specified set of abundances and/or ionization equilibria:
IDL > rad_loss,temperature,loss_rate
|
Given a set of observed spectral intensities, the problem is to invert a system of integral equations like the previous one. The procedure CHIANTI_DEM solves the system and calculates the DEM(T).
The inversion problem itself is not simple and requires some assumptions about the nature of the solution. A series of workshops was sponsored in 1990/91 to study differential emission measure techniques (Harrison and Thompson, 1992). It was found that most codes eventually gave consistent results, but that the DEM derived depends rather critically on the methods used to constrain the solution and the errors in the observed intensities and atomic data.
It is advisable to select a number of well resolved, unblended lines which are not density sensitive, emitted by various elements over a wide temperature interval. Appropriate values of the pressure (or density) and the elemental abundances must be chosen according to the region of the Sun being observed. The pressure value can be obtained once the values of the temperature and the density are estimated. To estimate the electron density the procedure CHIANTI_NE can be used. The temperature can be estimated for example using the procedure CHIANTI_TE.
The contribution functions C (T,lij,Ne) can be calculated using CHIANTI_DEM either at constant pressure or at constant electron density. It is also possible to vary the elemental abundances before starting the fit to deduce the DEM.
Many papers have been written on solar elemental abundances. See e.g. Meyer (1993) Widing and Feldman (1992), Mason (1992, 1995). A possible approach in determining elemental abundances is to use the detailed shape of the DEM distribution for ions from the same element and apply an iterative procedure to normalize the curves for different elements (e.g. Fludra and Shmelz 1995, Del Zanna et al. 1995).
The CHIANTI_DEM procedure
The main IDL routine which has been written to perform a differential emission measure analysis of EUV spectra using the CHIANTI atomic database is CHIANTI_DEM. Other procedures required to run CHIANTI_DEM are GET_CONTRIBUTIONS , DEM_FIT, ZION2SPECTROSCOPIC and Z2ELEMENT. The resulting DEM may then be used by other procedures to calculate a synthetic spectrum.
This package of routines will be replaced in the future by new more versatile versions.
The main inputs required by CHIANTI_DEM are :
1) the observed wavelength lobs [Å].
2) The observed flux Iobs in ergs cm-2 s-1 sr-1.
3) The corresponding error sobs on the flux in ergs cm-2 s-1 sr-1.
4) The value of dl [Å]. All the theoretical lines that may have contributed to the observed lines, i.e. that have a theoretical wavelength ltheo in a lobs 1dl range will be searched for. This value should correspond to the spectral resolution of the instrument at that wavelength.
5) The identification, written as a string of up to 20 characters. For example:
171.114 4811.0 1443.0 0.25 Fe IX
174.604 4005.0 1202.0 0.25 Fe X
180.448 3877.0 1163.0 0.25 Fe XI bl Fe X
195.149 3443.0 1033.0 0.25 Fe XII
Once the file with the observed fluxes is read, another IDL procedure, GET_CONTRIBUTIONS, is called by CHIANTI_DEM in order to calculate the contribution functions C(T,lij,Ne) at the given constant density or pressure.
GET_CONTRIBUTIONS searches the CHIANTI database for all the theoretical lines that may have contributed to the observed lines, i.e. that have a theoretical wavelength ltheo in a lobs 1dl interval.
Then, for each theoretical line selected, it calculates the C values for the temperature interval log(T)= 4.0 - 8.0 in steps of log(T) = 0.1.
If a constant pressure is selected, for each ion the contribution function is calculated at an electron density Ne equal to the ratio of the pressure and the temperature of maximum ionization fraction.
The C(T) values are stored by GET_CONTRIBUTIONS in the output file output.contributions that can be used later, if required, to re-calculate the DEM, changing various parameters (e.g. the abundances), without having to start again and read the CHIANTI database, which can take a long time.
The observed lines with no theoretical counterparts are automatically excluded. If this happens, you might consider starting again with a larger dl, to see if there are theoretical lines in the vicinity of the observed one.
Then you are asked to select an *.abund file present in the CHIANTI database or in the working directory, and eventually edit it, if you want to change some abundances.
The G(T) are calculated, multiplying each theoretical line by the abundance factor. The theoretical lines contributing to each blend are sorted by intensity and then their G(T) can be plotted if the keyword PLOT_GT was activated. It is recommended to do this the first time, to check if there are some observed lines which are heavily blended with lines of other elements. It might be better to exclude such lines in a second run.
The G(T) for each blend are then summed and plotted, and the calculation of the DEM starts, using the fitting routine DEM_FIT. A series of parameters can change the result (DEM), especially the number and position of the mesh points of the spline that represents the DEM. The fitting procedure is based on Bevington's Data Reduction and Error Analysis for the Physical Sciences. fortran programs.
The iteration is controlled using key-words (see below).
A series of outputs are created, all having extensions of the output name. For example, using test as the output name:
The abundance file, the ionization equilibrium file and the pressure used are written at the beginning.
Then there is one line for each observed line, with the identification present in the input file, the observed wavelength lobs, the observed flux Iobs, the calculated flux Itheo , the error on the flux sobs, the value ([(Itheo - Iobs)/(sobs )] )2 and finally the value of [(Itheo)/(Iobs)]
After this line, there is one line for each theoretical line contributing to the blend, with the identification, the theoretical wavelength ltheo, the configuration and terms, and the contribution (as a percentage) of each line in the blend to Itheo.
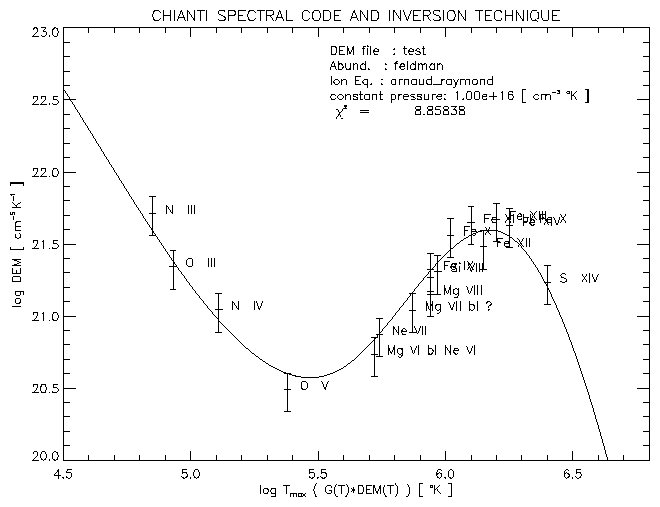
The first one has the G(T) of all the lines then used for the fit, with all the contributions for each line summed ( the labels refer to the identifiacation given in the input file).
The file test_dem.ps has the DEM (T) with the scales set as in the interactive session. The points are plotted at the temperature where the product G(T)*DEM(T) has a maximum. It is possible to label the points with the comment string present in the input file, or to use the dominant ion in the blend.
The file test_4plots.ps has some additional plots. The upper-right figure of test_4plots.ps plots the values [(Itheo)/(Iobs)] versus the temperature where G(T) has it's maximum.
The action of CHIANTI_DEM is controlled via the following keywords.
The default units are ergs cm-2 s-1 sr-1.
The default units are ergs cm-2 s-1 sr-1.
If set, it has to specify the name of the file previously created by GET_CONTRIBUTIONS, where all the contribution functions C(T) are stored.
There are also some actions controlled via the keyboard.
When you are asked for an answer ( [y/N] ) yes or no you should either type in y or n. The capital letter in [y/N] means that the default choice is n which is what you get if you simply hit the return key. In case you have [Y/n], hitting the return key is the same as choosing y .
You must specify the output file name and the value of the pressure (or the density). The input file name is optional.
IDL > CHIANTI_DEM,OUTPUT='test',FILE_INPUT='test_obs',PRESSURE=1e16,/PLOT_GT
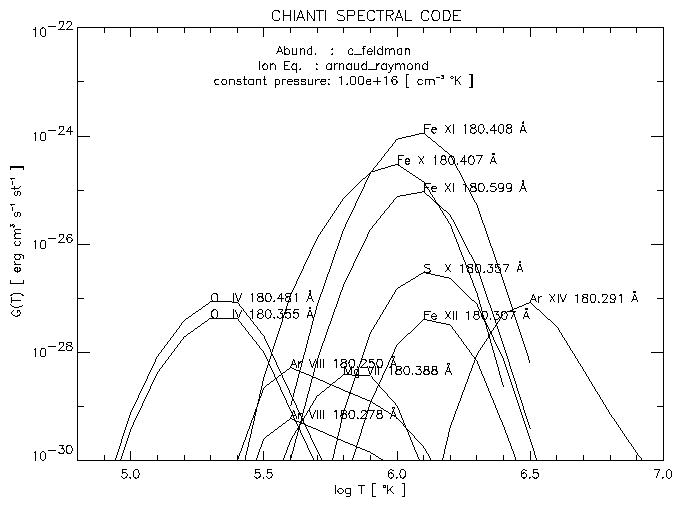
Select the ionization equilibrium file (e.g. Arnaud & Raymond). If there are no problems about N_MATCHES, the routine will select the lines having max( C(T) ) 3 10-30 and write the C(T) values to the file test.contributions.
Then you'll be asked to select an abundance file and if you want to edit it. Pick up the Feldman abundances. Then the G(T) are calculated, multiplying each theoretical line by the abundance factor, sorted (within each blend) by their max( G(T) ) value, and plotted ( see Fig. 8).
|
It is recommended that you check the plots at least once, to see if there are some observed lines that it might be better to exclude in a second run, for example because they are blends. Also check if your identifications are consistent with the lines found in the CHIANTI database.
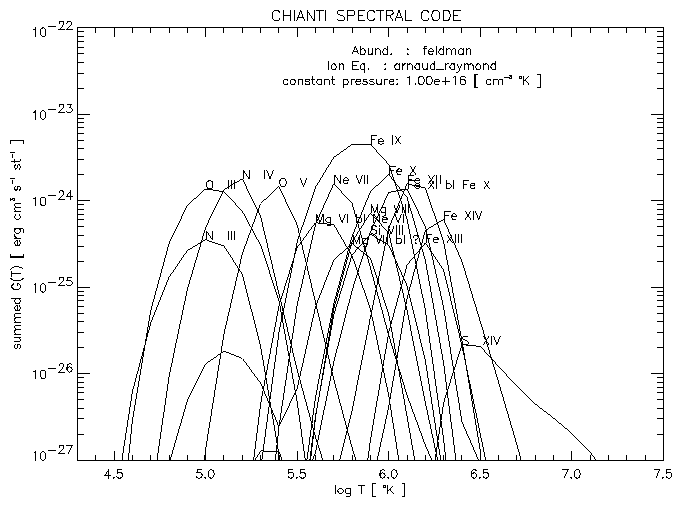
The G(T) for each blend are then summed, and plotted ( see Fig. 9).
|
At the end of the fit, the files test.dem, test.general, test.out are created.
Have a close look at these outputs, and check if there are emission lines not well represented by the fit or with no theoretical counterparts.
You can use the routine a second time, excluding some of the lines, and/or varying some of the fitting parameters. In particular, changing MESH_POINTS or starting from an appropiate DEM can affect the resulting DEM. For example:
IDL> CHIANTI_DEM,OUT='test',FILE_IN='test_obs',FILE_GT='test.contributions', $ IDL> EXCLUDE_OBS_WVL=[ 284.153 ] ,$ IDL > MESH_POINTS= [ 4.85, 5.6, 6.25, 7.0 ],N_ITER=40
The files test.dem, test.general, test.out will be created.
Eventually, also the files test_dem.ps test_4plots.ps may be created.
|
Fig. 10 shows the resulting DEM. The error bars on the points simply reproduce the error on the observed fluxes.
The Fig. 11 is self-explanatory. The DEM figure is repeated in the upper-left plot with the same scale of the previous plot.
The file test.out , toghether with test.dem, can be used to reproduce these plots using user-written software. If the only concern is the postscript output, then users just have to copy the routine in the working area and modify the top procedure PRINT2D_PLOT.PRO that controls the postscript device. The default is landscape.
This package is mostly intended to be a quick method to obtain a DEM which can then be used to calculate a synthetic spectrum, to be compared with the observed data.
Try to give as input lines covering a broad range in temperatures, and that are not density sensitive.
Try to adjust the location of the mesh points.
If the resulting DEM does not give a good fit to the data, it might be a good idea to start again calculating the G(T) with different abundances or to check the effect of blends.
Try a different DEM as a starting point, but be careful about the end points at lower and higher temperatures where usually there are no constraints (no observed lines).
Consider the possible effect on the DEM of different structures along the line of sight. It is important to realise that the DEM gives an indication of the amount of plasma at different temperatures along the line of sight, assuming constant density or pressure. It is not therefore possible to infer direct information about the variation of the temperature with height from this function. The inclusion of density-sensitive lines in the fit may also cause problems.
Send comments to : Giulio Del Zanna g.del-zanna@damtp.cam.ac.uk
The previous routines have been modified as described:
Compared to the previous version, these are the main changes:
1-Rewritten as a wrapper routine using the new procedures.
2-Now the PRESSURE value is a keyword.
3-The calculations can be done at constant DENSITY.
4-Energies (keV) can be output instead of wavelengths in Angstroms
5-MASTERLIST can now be used both as an input string or as a keyword.
Compared to the previous version, these are the main changes:
1-Rewritten as a wrapper routine using the new procedures.
2-Now the PRESSURE value is a keyword.
3-The calculations can be done at constant DENSITY.
4-MASTERLIST can now be used both as an input string or as a keyword.
Compared to the previous SYNTHETIC, these are the main changes:
1-Rewritten as a wrapper routine using the new procedures.
2-Now the PRESSURE value is a keyword as the DENSITY value
3-The keyword CONT is now renamed CONTINUUM
4-Added keywords
PHOTONS (If set, intensities are in photons instead of ergs)
DEM_NAME, ABUND_NAME, IONEQ_NAME, to run the routine in the
background, giving names of the
DEM, abundance and ionization
fractions files.
NOPROT (If set, then proton rates are not included.)
RADTEMP, RPHOT (to include photoexcitation)
5-MASTERLIST can now be used both as an input string or as a keyword.
6-The description of the line details now has the spectroscopic designation at the end.
Is left unchanged except for a few minor changes.
Aside from a few small fixes, these are the main changes:
1-Rewritten as a wrapper routine using the new procedures.
2-Added keywords
ABUND_NAME, IONEQ_NAME, to run the routine in the
background, giving names of the
abundance and ionization
fractions files.
NOPROT (If set, then proton rates are not included.)
RADTEMP, RPHOT (to include photoexcitation)
Compared to the previous routine, these are the main changes:
1-Rewritten as a wrapper routine using the new procedures.
2-Added keywords
ABUND_NAME, IONEQ_NAME, to run the routine in the
background, giving names of the
abundance and ionization
fractions files.
NOPROT (If set, then proton rates are not included.)
RADTEMP, RPHOT (to include photoexcitation)
EM Emission measure values.
Is not distributed nor supported anymore. Is now replaced by ch_ss.pro
Compared to the previous routine, these are the main changes:
1-Modified the plotting, adding information about the single lines.
2-Added keywords
NOPROT (If set, then proton rates are not included.) RADTEMP, RPHOT (to include photoexcitation)
Compared to the previous routine, these are the main changes:
1-Rewritten as a wrapper.
2-Added keywords
NOPROT (If set, then proton rates are not included.) RADTEMP, RPHOT (to include photoexcitation)
2-Added keywords
PHOTONS (If set, intensities are in photons instead of ergs) IONEQ_NAME The ionization fractions file. NOPROT (If set, then proton rates are not included.) RADTEMP, RPHOT (to include photoexcitation)
The call is the same, but the calculations are done using the fitting formulae of Itoh et al. (ApJS 128, 125, 2000) and Sutherland (MNRAS 300, 321, 1998). Also, a few bugs have been fixed.
Added the keyword
PHOTONS (If set, intensities are in photons instead of ergs)
-Added keywords
NOPROT (If set, then proton rates are not included.) RADTEMP, RPHOT (to include photoexcitation)
These routines are in the other/ subdirectory:
Unchanged.
Unchanged.
Widget unchanged but the low-level routine now uses emiss_calc.pro.
Unchanged.
Widget unchanged but the low-level routine now uses emiss_calc.pro.
Unchanged.
Changed the low-level routine that calculates the G(T), to account for the v.4 variations. The proton rates are included in the calculation of the level population.
Unchanged.
Routines in the extra/ subdirectory:
Is now a wrapper routine calling the widget RATIO_PLOTTER
Is now a wrapper routine calling the widget RATIO_PLOTTER
A few keywords have been added/ modified, mainly as an update for V.4.
A few keywords have been added/ modified, mainly as an update for V.4.
A few keywords have been added/ modified, mainly as an update for V.4.
A few keywords have been added/ modified, mainly as an update for V.4.
A few keywords have been added/ modified, mainly as an update for V.4.
This section describes the changes applied to the CHIANTI database and software in order to include the proton rates.
The proton rate data files have suffices .psplups and have the following format
2 3 2 0.000e+00 1.027e-02 8.000e+02-2.150e-13 1.052e-13 4.397e-12 \ 2.232e-11 5.389e-11 8.708e-11 1.014e-10 7.658e-11-2.805e-12
(The \ indicates a break in the line.) Note that the Z and spectroscopic number are not given for each ion, in contrast to the electron files. The rest of the line is the same as that for the electron files, except in this case there are 9 spline values as a 9-point spline was fitted to the data.
This is done by the routine read_splups.pro which loads the data into an IDL structure. The call is
IDL> read_splups_ns, filename, splstr, splref , /prot
The structure splstr has the following tags
.lvl1 lower level index
.lvl2 upper level index
.t_type transition type
.gf gf value
.de Delta-E for transition (rydbergs)
.c_ups the scaling parameter
.nspl number of spline points
.spl Vector of length 9, containing spline points
The routine pop_solver solves the set of linear equations that determine the level balance of the ion. To include proton rates, an extra matrix containing the proton rate coefficients needs to be added to the equations. This matrix is created within pop_solver from the splstr structure mentioned above.
Note also that the same routine is used to descale the spline fits into proton rates as to descale the electron spline fits. This routine is descale_all.pro and is called as
descale_all, temp, splstr, index, ups
where temp can be an array of temperatures.
User-written routines can be modified to include proton rates through two steps.
For routines which do not directly call pop_solver, the only changes are to add the keywords noprot=noprot, abund_file and ioneq_file to the routines' argument list, and to the call to the routine that calls pop_solver.
For routines that directly call pop_solver, the following steps need to be followed.
The first is to add the keyword noprot=noprot to the routine's argument list.
The second is to add a common block analogous to those that already exist for the energy level, radiative and electron collision data. This common block is
COMMON proton, pstr, pe_ratio
The third step is to add a line which will read the data. This is
if keyword_set(noprot) then begin
pstr=-1
endif else begin
read_splups, pname, pstr, pref, /prot
endelse
Before the call to pop_solver, the proton to electron ratio must be evaluated. This is done with
pe_ratio=proton_dens(temp)
where temp contains the logarithm of the temperatures that will be passed to pop_solver.
To include the proton rates in CHIANTI it is necessary to know the proton number density Np. This quantity is usually expressed in terms of the ratio relative to the electron density. For a standard solar plasma this is a constant for temperatures beyond log T=4.6 with a value around 0.85. Thus one option is to simply fix the ratio as a constant.
As we want CHIANTI to be applicable for low temperature plasmas, however, we have decided to explicitly calculate the ratio making use of the ion balance and abundance files, that uniquely determine Np. The relevant routine is PROTON_DENS.PRO. Np is calculated from the ion balance and element abundance files contained in CHIANTI through the following expression
| (24) |
where Ab is the element abundance, Ai is the ith element (i.e., A1=H, A2=He, etc.), Z is the charge on the ion, F(Ai+Z, T) is the fraction of ions of element Ai in the form Ai+Z at temperature T.
The ion fractions contained in CHIANTI are tabulated over the range 4.0 # log T # 8.0. Above and below these values, we set R(T) to the values for log T=8.0 and log T=4.0, respectively.
The use of this routine has some side effects. Some routines for which the ratio may have some effects in the results don't require you to select the ion balance or abundance files. E.g., DENSITY_RATIOS.PRO does not require the user to select these files, however, at low temperatures one may see significant changes take place in line ratios on account of the change in the proton-to-electron ratio. We deal with this effect by using the default !abund\_file and !ioneq\_file files to derive the proton-to-electron ratio, but allowing the files to be directly specified by the user through keywords if he/she needs to do this.
We have modified the CHIANTI routines so as PROTON_DENS.PRO is called once at the beginning of the routine and the ratio data are passed to POP_SOLVER.PRO through a common block.
The modifications due to the proton-to-electron ratio in user-defined procedures are as follows:
common proton, pstr, pe_ratio
COMMON elements,abund,abund_ref,ioneq,ioneq_t,ioneq_ref
pe_ratio=proton_dens(temp)
The call to PROTON_DENS.PRO thus does not take place within POP_SOLVER.PRO
This section describes the changes in the IDL software required to incorporate the 9-point spline fits.
The modification to the .splups file is simple. Any transitions which are fitted with a 9-point spline will simply have four extra numbers placed on the end of the data line. Transitions fitted with a 5-point spline will be output as normal. Both 5 and 9-point fits can be found in the same .splups file.
This section describes the changes to the read_splups routine, and also the changes required to routines that call directly or indirectly read_splups.
For some routines (such as synthetic, ...?) the reading of the data files is delegated to the routine setup_ion.pro. The data is then transferred to the main routine via common blocks.
Every routine that makes a call to read_splups.pro has to be modified. Two changes are needed:
The read_splups call used to be
read_splups,upsname,t_type,gfu,deu,c_ups,splups,upsref
The new call is
read_splups, filename, splstr, splref [, /prot]
where /prot should be set if proton rates are to be read.
The original common block had the form
COMMON upsilon,t_type,deu,c_ups,splups
The new one should be
COMMON upsilon, splstr
This reads data from .splups or .psplups files into an IDL structure (the previous version read data into several individual arrays). The call is
IDL> read_splups, filename, splstr, splref [, /prot]
The structure splstr has the following tags
.lvl1 lower level index
.lvl2 upper level index
.t_type transition type
.gf gf value
.de Delta-E for transition (rydbergs)
.c_ups the scaling parameter
.nspl number of spline points
.spl Vector of length 9, containing spline points
This routine directly replaces descale_ups.pro in the CHIANTI software. It is called as
descale_all, temp, splstr, index, ups
An important difference is that the temperature is specified directly whereas previously the scaled temperature was given. In addition, several temperatures can be given at once rather than just one.
Note that there is no difference in how this routine treats the descaling of proton rates.
The only place in the CHIANTI software where this routine is called is in pop_solver.pro.
The way electron excitation rates are included in pop_solver has been changed. Only a single for loop is required now as the routine goes through each line in the structure. In addition, the routine can now descale the upsilons for several temperatures simultaneously.
This section describes the features of the routines that are contained in the extra/ directory and that were contributed by Peter Young:
These routines, described in more detail in Sect. D.2 below, have slightly different units of outputs, compared to the other routines.
First of all, Sect. 6 gives the theory
behind the interpretation of optically-thin emission lines which
serves to set out the notation used here.
Going back to Eq. 2, we write
| (25) |
The emissivity of the emission line resulting from a j-to-i
radiative decay is defined as
| (26) |
| (27) |
In order to relate the emissivity to the actual observed intensity of
a line, we make use of the third assumption, which tells us that the
intensity is proportional to the emissivity of the plasma, and so
| (28) |
Expanding el using Eqs 25 and 26 gives
| (29) |
| (30) |
| (31) |
On account of the ionisation fraction F(T) this function is sharply peaked, and a common approximation (e.g., Pottasch [3], Jordan & Wilson [1]) is to assume that G(T) has a constant value over a narrow temperature interval around G(Tmax), where Tmax is the temperature of maximum ionisation for the ion. Here we will use the temperature of maximum emission or Tmem which is the temperature at which Gl has its maximum. Defining
| (32) |
| (33) |
| (34) |
| (35) |
| (36) |
Now, solar emission lines are often measured as intensity (or radiance), I, with units typically of erg cm-2 sr-1 s-1. This quantity is related to Pl by
| (37) |
where dA is the projected area of the emitting element. One thus relates the observed intensity to an emission measure by
| (38) |
where EM(s) is the column emission measure, where s is the line-of-sight depth of the emitting region.
Stellar emission lines are measured in flux (or irradiance), E, with units typically of erg cm-2 s-1. E is related to Pl by
| (39) |
where d is the distance to the object. The observed flux is then related to the emission measure by
| (40) |
If one treats the emitting region as a uniform, spherical shell of thickness h then dV=4pR2dh (R the distance from the star centre of the shell; typically R=R*, the radius of the star) and so the expression for E becomes
| (41) |
where EM(h) is the emission measure over height. The factor 1/2 denotes that half the photons from the shell are emitted towards the stellar surface and so are destroyed. Jordan and co-workers (see, e.g., Jordan & Wilson [1], Jordan & Brown [2]) utilise this definition and an assumption of spherical symmetry to deduce energy balance relations in solar and stellar atmospheres.
The routines are divided into primary and secondary routines. The secondary ones are called by some of the primary routines, and chances are that you won't have to use them too often. They are described in Sect. D.3.
All of the routines have headers which give more detailed information about how they work. This header can be read in the normal IDL way through, e.g., doc_library,'ratio_plotter'.
| (42) |
Example: For Fe XIII, select a line/blend from lines
in the range 200 to 205 Å
pop_plot,26,13,wrange=[200,205]
Note how no single line shows zero density dependence, and so care should be taking in using Fe XIII in emission measure analyses. Compare with Fe XVI:
pop_plot,26,13,wrange=[330,370]
where both the 335 and 360 lines are OK.
This routine calculates Cl, defined in Eq. 34. It displays both this value and the values of DE Cl and 4p/ DE Cl. For lines for which njAji ~ Ne, Cl is insensitive to Ne, but for other lines Ne should be specified. Note that for blended lines only eDE Cl and 4p/ eDE Cl are output. The routine also outputs the Tmem of the lines, accurate to 0.02 dex.
Example: Work out Cl for the Fe XIII lines
between 200 and 205 Å at a density of 109 cm-3.
integral_calc,26,13,wrange=[200,205],dens=9.
From Eq. 38, an observed line intensity of 100 erg cm-2s-1sr-1 for the 202.044 line implies a column emission measure of EM(s) = 100 ×1.614 ×1020 / Ab(Fe), where 1.614 ×1020 is taken from 4pi/DE*C_lambda column of the output.
For Fe XIV, one can do:
integral_calc,26,14,wrange=[210,220],dens=9.
and so to get the same column emission measure for Fe XIV l211.32, an intensity of 100 ×1.614 ×1020 / 2.280 ×1020 = 70.8 erg cm-2s-1sr-1 is required, where 2.280 ×1020 is the value of 4pi/DE*C_lambda for Fe XIV l211.32.
Both temp\_plotter.pro and dens\_plotter.pro call a widget-based routine (ratio_plotter, via the keywords /temp and /dens) that allows the thorough investigation of density or temperature sensitive ratios. Observed line intensities can be input for line ratios, and densities or temperatures derived.
Example: to study density sensitive ratios of Fe XIII, simply
type in
dens_plotter,'fe_13'
Try inputting some line intensities and errors from the SERTS-89 spectrum (Thomas & Neupert [4]), and comparing the derived densities with those found by Young, Landi & Thomas [5] in Table 20.
Gives percentage level populations for all levels within the specified ion that have populations greater than 0.01%.
Example: Compute level populations for Fe XIII at a
density of 1010 cm-3:
show_pops,26,13,dens=10
Eq. 31 gives the definition of the
contribution function as calculated by the g_of_t
routine.
In it's default setting g_of_t.pro actually calculates:
|
Examples:
result=g_of_t(26,13,dens=9.)
result=g_of_t(26,13,wrange=[200,205],/abund)
result=g_of_t(26,13,/no_de)
One can also use this routine to derive the Tmem of the emission line, by way of the ion_interp.pro routine, e.g.,
result=g_of_t(26,13,dens=9.)
ion_interp,t,result,ti,g_ti,10
print,ti(where(g_ti eq max(g_ti)))
result is tabulated at 0.1 dex intervals in temperature. ion_interp interpolates result and in this case gives it at 0.01 dex intervals.
These routines are called by the routines above.
Calculates the ion emissivity (Eq. 27) for all transitions within the CHIANTI model of the ion. The returned data is in the form of a structure. The default is to calculate emissivities for temperatures Tmax and log Tmax10.15, and densities log Ne=8.0, 8.5, 9.0,....,12.0.
Example:
emiss=emiss_calc(26,13)
Allows the selection of lines/blends from the emiss structure created by emiss_calc.pro. This routine is useful if you want to access the emissivities of lines directly, e.g.,
emiss=emiss_calc(26,13) em202=emiss_select(emiss,wra=[200,205],sel_ind=sel_ind)In this example, calling emiss_select yields a widget that allows one to select a line/blend from the 200-205 Å range. The emissivities of this line blend will be contained in em202, while the emiss index/indices of this line/blend will be contained in sel_ind.
When reading the ionisation equilibrium files, you will receive an array with absolute (as opposed to log) ion fractions tabulated at 0.1 dex intervals from log T = 4.0 to 8.0. A common need is to interpolate this data and obtain the ion fraction tabulated at smaller intervals. As the ion fractions are generally sharply peaked, normal interpolation will lead to negative ion fractions at several temperatures, and so a more satisfactory method is to interpolate the log of the ion fraction. However, you need to take the log of only the non-zero values of the ion fraction.
The several lines of code required to perform the interpolation are straightforward but irritating (when typed on many occasions!), and so this routine performs the task.
Example: Use g_of_t to create a G(T) function for
one of the Fe XIII lines,
result=g_of_t(26,13,dens=9.)
ion_interp,t,result,ti,g_ti,5
The G(T) function is now tabulated at 0.02 dex intervals. Note that if t is not specified, it is assumed to be a vector going from 4.0 to 8.0 in 0.1 dex intervals.
The calling sequence is:
IDL> ch_synthetic, wmin, wmax, output=output, err_msg=err_msg, msg=msg, $
pressure=pressure, density=density, $
model_file=model_file, all=all,sngl_ion=sngl_ion, $
photons=photons, masterlist=masterlist, $
save_file=save_file , verbose=verbose,$
logt_isothermal=logt_isothermal, logem_isothermal=logem_isothermal,$
goft=goft, ioneq_name=ioneq_name, dem_name=dem_name, $
noprot=noprot, rphot=rphot, radtemp=radtemp, progress=progress
PROCEDURE:
This routine calculates as default line intensities for a user-specified
differential emission measure and ionisation balance. The actual
creation of a synthetic spectrum (i.e., wavelength vs. intensity)
is performed by other routines - see CH_SS.PRO and
MAKE_CHIANTI_SPEC.PRO.
Note that this routine does not include the element abundances
in the line intensities, as this will be performed by
make_chianti_spec. One of the reasons why element abundances are not
included in the line intensities calculation is so that it is easier
for the user to see how modifying abundances affects their spectra in
e.g. CH_SS.PRO.
The calculations are performed at constant pressure or
at constant density.
The routine can also output line intensities calculated with an
isothermal approximation.
If the isothermal approximation is not used, then the user will be asked
to select two files, that can either be in the
standard CHIANTI database or in the working directory.
These files are:
- an ionization fraction file
- a differential emission measure (DEM) file.
The routine can also output the contribution functions G(T) of the lines,
instead of the intensities, if the keyword GOFT is used. In this case,
only the ionization equilibrium file needs to be selected.
The G(T), or intensity per emission measure, is calculated as:
G=(hc/lambda_ij)*A_ji*(N_j(X^+m)/N(X^+m))*(N(X^+m)/N(X))/ N_e /(4.*!pi)
where A_ji is the A-value of the transition
(N_j(X^+m)/N(X^+m)) is the population of the upper level,
calculated by solving the statistical equilibrium equations
(N(X^+m)/N(X)) is the ionization equilibrium
N_e is the electron density.
unless /PHOTONS is set, in which case the (hc/lambda_ij) factor
is not included.
If not specified otherwise, with the use of the MASTERLIST or SNG_ION
keywords, then the standard masterlist of the ions, which has
all the ions in the current CHIANTI database, is used.
PROGRAMMING NOTES
The DEM is not assumed to be specified at 0.1 logT intervals (which
is how the ion fraction are specified). Thus this routine reads
in the DEM vs. logT information and then uses the IDL spline
function to tabulate the DEM over 0.1 logT intervals. The minimum
and maximum temperatures are those in the DEM file, rounded up to
the nearest 0.1. The new DEM function tabulated over 0.1 logT
intervals is contained in 'dem_int'.
For some of the dielectronic files, radiative decays that were in
the standard .wgfa file will also be present in the dielectronic
version of the .wgfa file. In these cases the line intensity
produced from the latter file needs to be ignored and so we have a
check in ch_synthetic to do this. An example is the 1-7 decay in
the ca_19.wgfa and ca_19d.wgfa files. In the latter case, the
model of the ion does not include electron excitation to level 7
and so the model for the 1-7 decay is incorrect, hence we ignore
it.
INPUTS:
Wmin: minimum of desired wavelength range in Angstroms
Wmax: maximum of desired wavelength range in Angstroms
PRESSURE: pressure in emitting region (Pe, cm^-3 K).
Only a single value is accepted, and the calculation is
performed at constant pressure.
OPTIONAL INPUTS :
DENSITY: density in emitting region (Ne, cm^-3).
Only a single value is accepted, and the calculation is
performed at constant density, unless LOGT_ISOTHERMAL is
defined. In this case, DENSITY can be an array of values, but
has to have the same number of elements as LOGT_ISOTHERMAL.
MODEL_FILE Full path of the (Te,Ne) file if defined.
This file should have two columns, one with the Te (K)
values, and one with the Ne (cm^-3) values. If these
values are not sorted in ascending order of Te, the
routine does sort them.
SNGL_ION: specifies a single ion (e.g. SNGL_ION='Fe_10' to include
only Fe X lines) or an array (e.g. SNGL_ION=['Fe_10','Fe_11']
to include only Fe X and Fe XI lines) of ions to be used
instead of the complete set of ions specified in
!xuvtop/masterlist/masterlist.ions
MASTERLIST: string of a specific masterlist file (full path).
If defined as a keyword (i.e. MASTERLIST=1 or /MASTERLIST)
then a widget allows the user to select a user-defined
masterlist file. Shortcut for SNGL_ION.
IONEQ_NAME: Name of the ionization equilization name to use. If not
passed, then the user is prompted for it.
DEM_NAME: Name of the DEM file to used. If not passed, then the user
is prompted for it.
LOGT_ISOTHERMAL
Array of logarithmic temperatures.
If defined, the emissivities are calculated with an
isothermal approximation. The values are sorted in ascending
order.
LOGEM_ISOTHERMAL
Array of logarithmic emission measures.
If defined, the emissivities are calculated with an
isothermal approximation. The values are sorted in ascending
order. If LOGT_ISOTHERMAL is specified without
LOGEM_ISOTHERMAL then the emission measures are set to 1
(logem_isothermal=0).
RADTEMP The blackbody radiation field temperature (default 6000 K).
RPHOT Distance from the centre of the star in stellar radius units.
I.e., RPHOT=1 corresponds to the star's surface. (Default is
infinity, i.e., no photoexcitation.)
OUTPUTS:
OUTPUT: The name of the structure containing the line intensities and
details.
OPTIONAL OUTPUTS:
SAVE_FILE: If defined, then an IDL save file is created, with the output
structure.
GOFT: If set, the G(T) of the lines are calculated, and put in
the output structure, instead of the line intensities.
KEYWORDS:
ALL: if set, then all lines are included. This means that lines for
which only an approximate wavelength is known,
denoted by a negative wavelength value in the .wgfa file, are
included. These are the lines for which there are no observed
energy levels.
PHOTONS: The output intensities will be in photons instead of
ergs.
VERBOSE: If set, the routine will list each ion it is looking at,
and how many lines from each ion it is including in the
spectrum.
GOFT: If set, the G(T) of the lines are calculated, and put in
the output structure, instead of the line intensities.
NOPROT Switch off the inclusion of proton rates in the level
balance (default).
PROGRESS If set, a widget appears, showing the progress of the
calculation and allowing the user to halt the calculation.
NO_SUM_INT Prevents the summing of intensities over temperature.
Only works in conjunction with the LOGT_ISOTHERMAL
option, and is implemented in order to work the
ISOTHERMAL routine. The .INT tag in OUT.LINES becomes
an array with the same number of elements as
LOGT_ISOTHERMAL, corresponding to the intensities at
each temperature.
The tags of the line intensities structure are:
.lines A structure containing information about the lines.
Its size is the number of lines in the spectrum. The
tags are:
.iz The atomic number of the elements (e.g., 26=Fe)
.ion The ionisation stage (e.g., 13=XIII)
.snote The identification of the ion (e.g., 'Fe XXIV d')
.ident The identification of the transition, configuration
and terms in text form.
.ident_latex
The identification of the transition, configuration
and terms in latex form.
.lvl1 The lower level of the transition (see .elvlc
file for ion)
.lvl2 The upper level for transition.
.tmax The temperature of maximum emission of the line.
If the G(T) are output, tmax is the maximum of G(T).
If the isothermal approximation is used tmax=0.
If a DEM is used, tmax is the maximum of the
emissivity that includes the product of the ion
fraction and the DEM.
Rounded to nearest 0.1
.wvl Wavelength of the transition, in Angstroms.
.flag A flag, =-1 if the line has only theoretical energy
levels. Otherwise flag=0.
.int Intensity of line (erg/cm2/s/sr or phot/cm2/s/sr),
divided by the element abundance (exclusive with .goft).
.goft The G(T) of the line (optional /exclusive with .int).
.ioneq_name The ion balance file used (full path).
.ioneq_logt The Log10 T values associated.
.ioneq_ref The references.
.dem_name The differential emission measure file eventually used
(full path).
.dem The Log10 DEM values
.dem_logt The Log10 T values associated.
.dem_ref The references.
.model_name A string indicating the model used:
1- Constant density
2- Constant pressure
3- Function (Te,Ne)
.model_file Full path of the (Te,Ne) file if defined. Null string otherwise.
.model_ne the Ne value(s).
- a scalar if 'Constant density' is selected.
- an array if 'Function' is selected.
- 0. if constant pressure is selected.
.model_pe the Pe value.
- a scalar if constant pressure is selected.
- 0. if 'Constant density' is selected.
- an array=density*temperature if 'Function' is selected.
.model_te the Te values if 'Function' is selected. Otherwise 0.
.wvl_units The wavelength units.
.wvl_limits The wavelength limits specified by the user.
.int_units The intensity units (erg/cm2/s/sr or phot/cm2/s/sr) if
intensities are calculated, otherwise the G(T) units
(erg cm3/s/sr or phot cm3 /s/sr)
.logt_isothermal
The Log10(T) values used.
.logem_isothermal
The Log10(EM) values used.
.date The date and time when the structure was created.
.version The version number of the CHIANTI database used.
.add_protons
A flag (0/1) to indicate whether proton data were used (1)
or not (0) to calculate the level population.
.photoexcitation
A flag (0/1) to indicate if photoexcitation was included (1)
or not (0).
.radtemp
The blackbody radiation field temperature used (if
photoexcitation was included).
.rphot
Distance from the centre of the star in stellar radius units
(if photoexcitation was included).
The calling sequence is:
IDL> ch_line_list, transitions, outname, latex=latex, ascii=ascii, $
wmin=wmin,wmax=wmax,$
SPECTRUM=SPECTRUM, abundfile=abundfile, min_abund=min_abund, $
minI=minI,photons=photons,kev=kev, $
all=all,no_sort=no_sort, sngl_ion=sngl_ion
INPUTS:
The OUTPUT structure created by CH_SYNTHETIC
OPTIONAL INPUTS:
Wmin: lower limit of the wavelength/energy range of interest (Angstroms)
if kev keyword set, then wmin is in kev
Wmax: upper limit of the wavelength/energy range of interest (Angstroms)
if kev keyword set, then wmax is in kev
Mini: Minimum intensity for line to be included in output
SNGL_ION: specifies a single ion (or a list of ions) to be used instead
of the complete set of ions specified in the structure.
MIN_ABUND: If set, outputs only those elements which
have an abundance greater than min_abund.
For example, from Allen (1973):
abundance (H) = 1.
abundance (He) = 0.085
abundance (C) = 3.3e-4
abundance (O) = 6.6e-4
abundance (Si) = 3.3e-5
abundance (Fe) = 3.9e-5
KEYWORD PARAMETERS:
LATEX: Create a latex file (default, exclusive with /ASCII)
ASCII: Create an ascii file (exclusive with /LATEX)
MINI: Minimum intensity for line to be included in output
PHOTONS: units will be in photons rather than ergs
KEV: wavelengths will be given in kev rather than Angstroms
ALL: if set, then all lines are included. This means that lines for which
only an approximate wavelength is known, denoted by a negative
wavelength value in the .wgfa file, are included.
These lines are listed in the file with a * preceding the wavelength.
NO_SORT:
If set, then the lines are *not* sorted in wavelength (or energy).
SPECTRUM
If set, IT IS ASSUMED that the input structure is the SPECTRUM
structure output of MAKE_CHIANTI_SPEC, where the line
intensities have already been multiplied by the abundance factor!!
OUTPUTS:
A latex (default) or an ascii file OUTNAME with the line list
The calling sequence is:
IDL> make_chianti_spec, TRANSITIONS, LAMBDA, OUTPUT, BIN_SIZE=BIN_SIZE, $
INSTR_FWHM=INSTR_FWHM, BINSIZE=BINSIZE, $
WRANGE=WRANGE, ALL=ALL, continuum=continuum, $
ABUND_NAME=ABUND_NAME, MIN_ABUND=MIN_ABUND, $
photons=photons, file_effarea=file_effarea, $
err_msg=err_msg, verbose=verbose
INPUTS :
TRANSITIONS, the structure created by ch_synthetic.pro.
OPT. INPUTS :
LAMBDA Array of wavelengths (X-values). If not defined as input, it is
calculated on the basis of BIN_SIZE, and returned as an output.
If defined as input, the routine checks that there are at least
10 points in the wavelength range defined by WRANGE. If there
are, the corresponding subset of LAMBDA is returned, otherwise
the routine exits with an error.
BIN_SIZE Bin size in Angstroms of the spectrum to be created. A linear
spectrum is assumed. In case an effective area file is used, the
wavelenghts in the file (that might not be linear) are used to
create the spectrum, and this bin size looses any meaning.
WRANGE Allows a subset of the wavelength range to be turned into
a spectrum. When using syn_plot, this speeds up the routine
a lot if you've elected to zoom in on a region.
INSTR_FWHM Instrumental FWHM (Angstroms).
In case an effective area file is used, The line intensities
contributing to each bin are summed, and subsequently convolved
with a gaussian of full-width-half-maximum FWHM if FWHM is not
set = 0 . Please make sure that the FWHM value (if not set to
zero) is larger than the bin size.
ABUND_NAME A CHIANTI abundance file name can be set.
It allows the abundance file given in transitions.abund_name
(if present) to be over-ridden. Note that it also used for
the continuum calculation.
MIN_ABUND: If set, calculates line intensities only from those elements
which have an abundance greater than min_abund.
Can speed up the calculations. For example, from Allen
(1973):
abundance (H) = 1.
abundance (He) = 0.085
abundance (C) = 3.3e-4
abundance (O) = 6.6e-4
abundance (Si) = 3.3e-5
abundance (Fe) = 3.9e-5
FILE_EFFAREA
The Effective Area File (TWO COLUMNS - wavelengths in
Angstroms and cm^2 values ) If defined, then the spectrum is
multiplied with these values. Any input LAMBDA value is
over-written by the wavelenghts in the file (that might not
be linear and are used to create the spectrum.
Note that this option only works well if a sufficient number
of bins is given. The line intensities contributing to each
bin are summed, and subsequently convolved with a gaussian
of full-width-half-maximum FWHM, if FWHM is not set = 0.
Please note that the convolution might not work if a small
number of bins is defined.
OUTPUTS :
LAMBDA Array of wavelengths (X-values).
If not defined as input, it is
calculated on the basis of BIN_SIZE, and returned as an output.
If defined as input, the routine checks that there are at least
10 points in the wavelength range defined by WRANGE. If there
are, the corresponding subset of LAMBDA is returned, otherwise
the routine exits with an error.
SPECTRUM A CHIANTI spectrum structure containing all
the information (see below).
OPT. OUTPUTS:
BINSIZE If BIN_SIZE is not specified, then the spectrum
bin-sizes are computed automatically, and the size of the
bin returned in BINSIZE.
KEYWORDS :
PIXEL The spectrum is given in /pixel units rather /ang
(DISABLED)
ALL Add lines that originally had negative wavelengths
PHOTONS If set=1, the output intensities will be in photons instead of
ergs.
CONTINUUM
If set, then the continuum is added to the
spectrum.
verbose
RESTRICTIONS: The input structure has to be of the type created by ch_synthetic.
The LAMBDA, EFFAREA values must be ordered in wavelength and the
LAMBDA values must be in Angstroms.
The spectrum structure output of MAKE_CHIANTI_SPEC has the following ADDITIONAL tags (compared to the tags of the CHIANTI line intensities structure created by CH_SYNTHETIC:
LAMBDA The array of X-values
SPECTRUM The array of Y-values
UNITS The units of LAMBDA, SPECTRUM
WRANGE The wavelength range
INSTR_FWHM The Instrumental FWHM
BIN_SIZE Width of the Bins (fixed) in angstroms
ABUND_NAME The CHIANTI abundance file name
ABUND The abundance values
MIN_ABUND The minimum abundance value used
ABUND_REF The references
CONTINUUM The values of the continuum (if calculated)
FILE_EFFAREA The Effective Area File used (optional)
EFFAREA The array of effective area values
(optional - same size of LAMBDA)
.LINES An array of structures, for all the lines used
to calculate the SPECTRUM.
The tags are the same as those created by
CH_SYNTHETIC, plus
.PEAK The peak intensity of the line in the spectrum
(approx. value)